Introduction to Metagenomics
Why study the microbiome? Amplicon and shotgun sequencing. Analysis pipelines and Visualisation options
Why study the microbiome?
Health care research
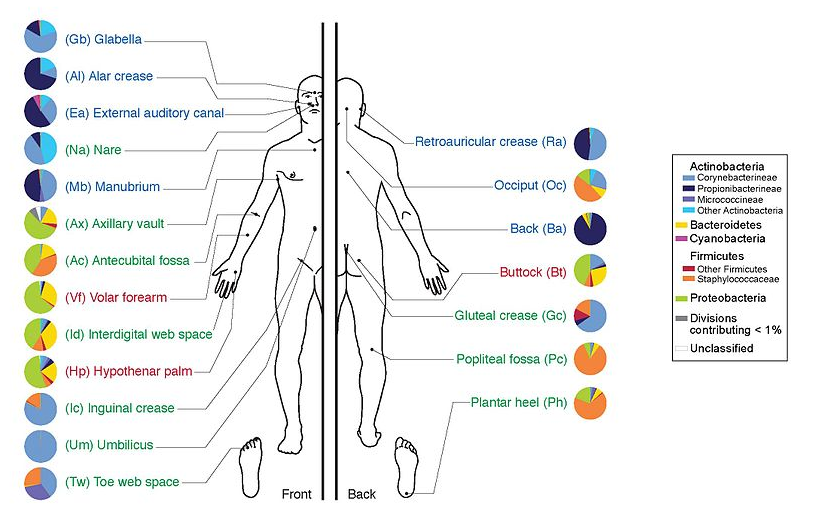
- Humans are full of microorganisms
- Skin, gut, oral cavity, nasal cavity, eyes, ..
- Affects health, drug efficacy, etc
- Sometimes referred to as your second genome
- ~10 times more cells than you
- ~100 times more genes than you
- ~1000s different species
Go Up
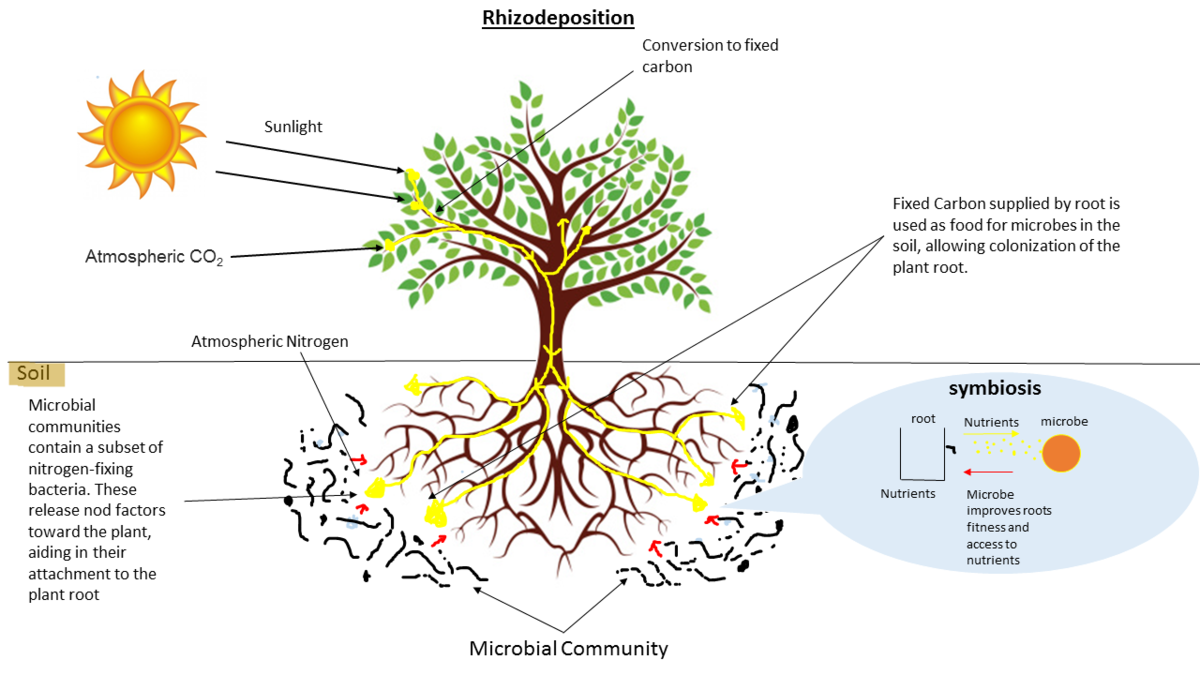
Environmental studies
- Microbes in the soil affect plants and animals
- Improve agriculture
Go Up
2 different approaches
Puzzle analogy: think of each organism as a puzzle
Shotgun
We consider all pieces (DNA) from all puzzles (organisms); we throw the pieces in a big pile, and need to reconstruct each individual puzzle from this. It is more complex, but you get full details (functional information) about the picture on each puzzle.

- Sequence all DNA
- More information
- Higher complexity
- Higher cost
Go Up
Amplicon
We only look at the corner pieces, these are easy to spot, and all puzzles have them. It will not tell us all the details (e.g. functional information) of the picture on the puzzle, but might be enough to tell us whether it is a landscape or a portrait or abstract art for instance (taxonomy).

- Sequence only specific gene
- No functional information
- Less complex to analyse
- Cheaper
Go Up
- Targeted approach, e.g. 16S (bacteria), 18S (eukaryotes), ITS (fungi)
- Amplify gene of specific taxonomic group, i.e. not host or environment
Go Up
- Highly conserved gene: easy to target across all bacteria
- With variable regions: distinguish between genus
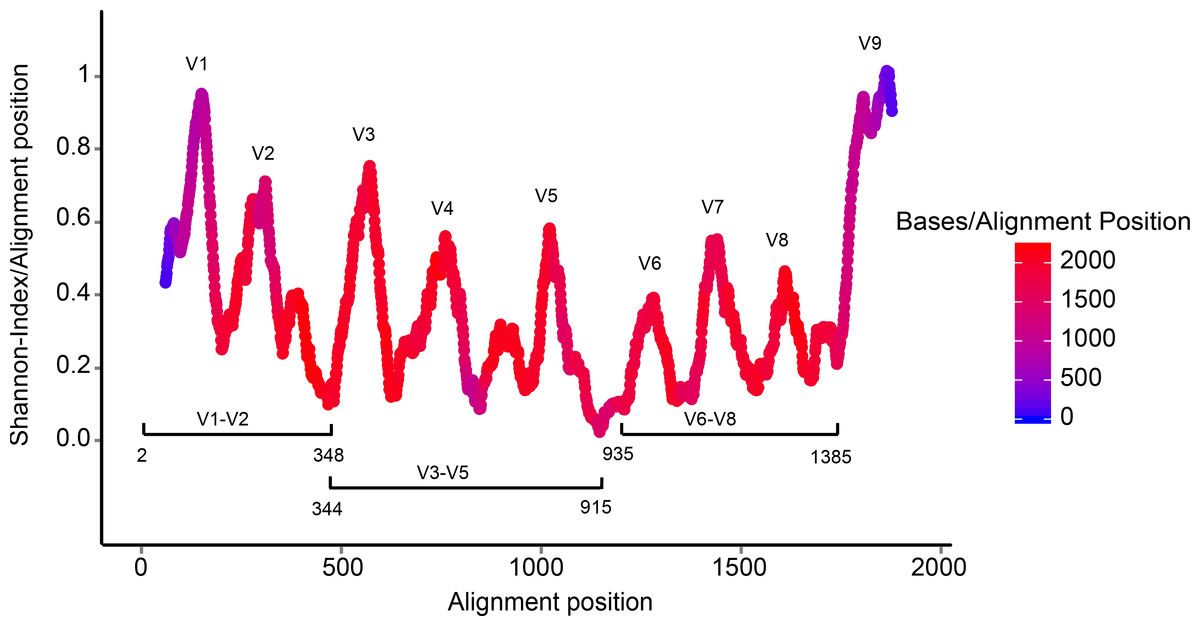
V1, V2 etc are the variable regions
Go Up
- Pros
- Well-established
- Inexpensive ($50-$100/sample)
- Cons
- V-region choice can bias results
- Is based on a very well conserved gene, making it hard to resolve species and strains
Go Up
Aims to sequence the “whole” metagenome
- Pros:
- Not biased by amplicon primer set
- Not limited by conservation of the amplicon
- Can also provide functional information
- Cons:
- Environmental contamination, including host
- More expensive ($1000+/sample)
- Complex data analysis
- Requires high performance computing, high memory, high compute capacity
Go Up
Analysis pipelines
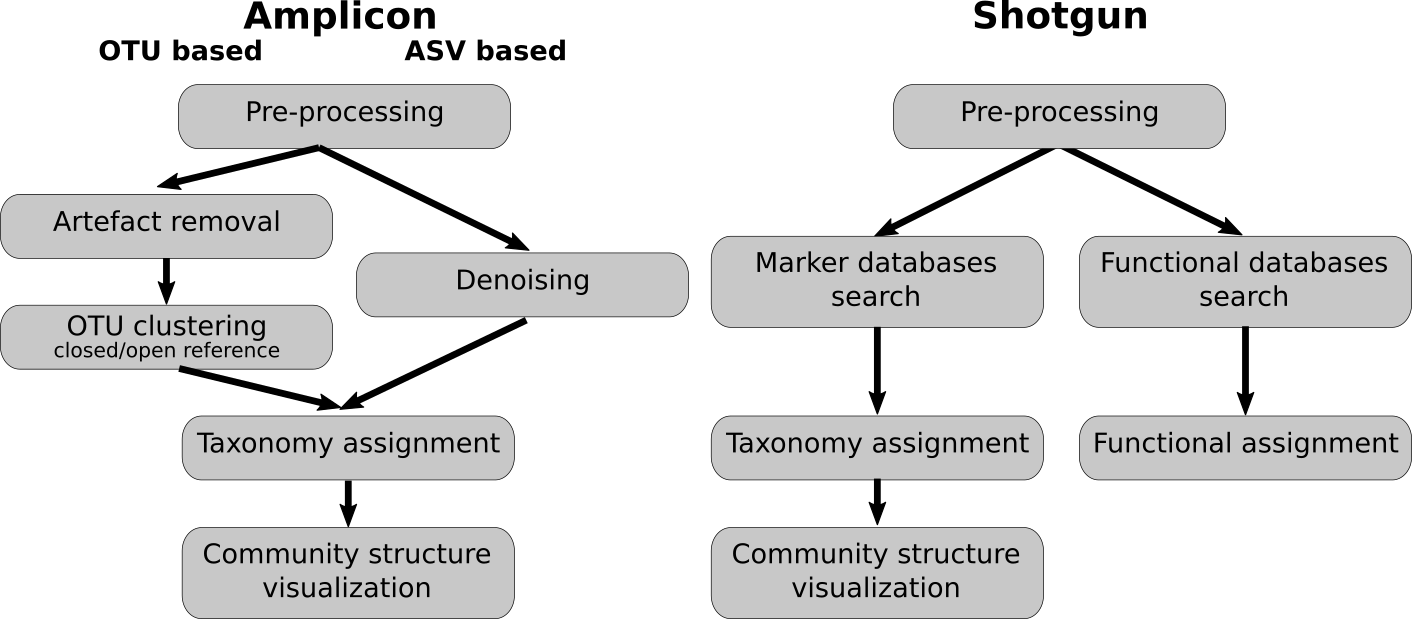
- OTU: operational taxonomic units
- ASV: amplicon sequence variants
Go Up
Pre-processing
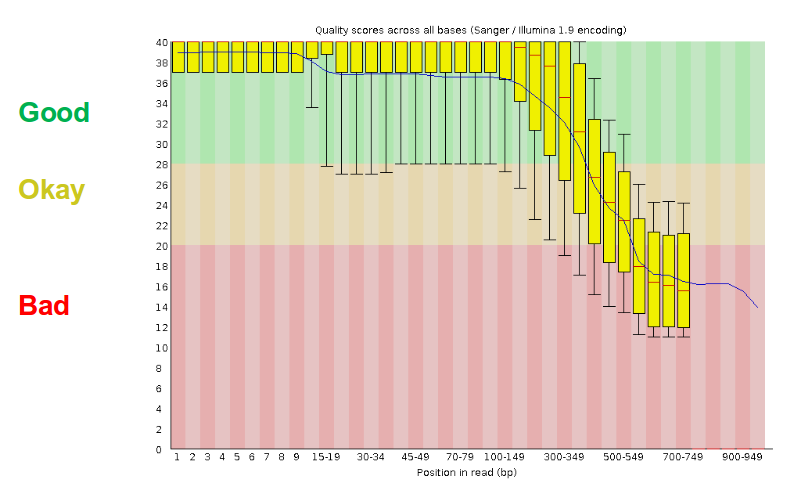
- There are a lot of ways to filter and trim your data
- Trade-off between quality and amount of information retained
Go Up
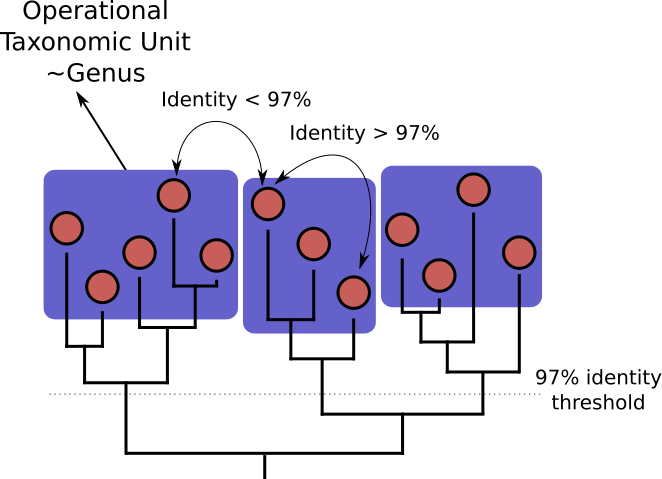
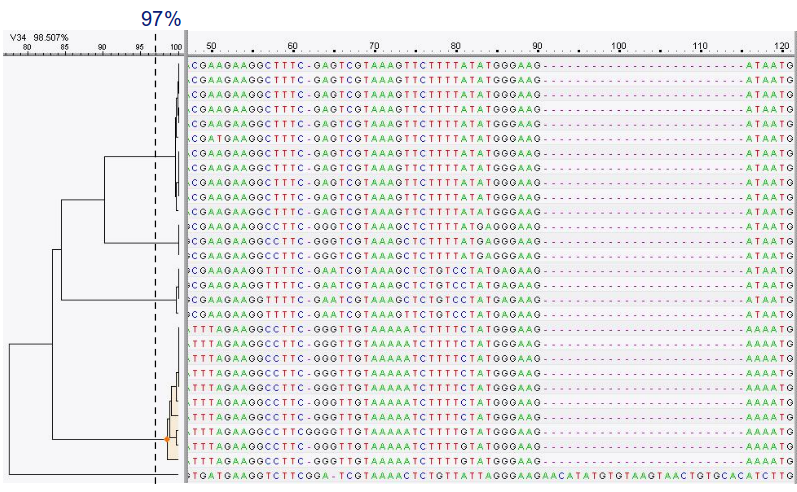
OTU Clustering
Operational Taxonomic Unit (OTU) clustering is a method used to group similar sequences of DNA together
into operational taxonomic units.
When clustering the dataset, OTUs will be generated by combining sequences that are similar to a set percentage level
(traditionally 97%), with the most abundant sequence identified as the true sequence.
- Cluster on 97% sequence similarity for genus-level differentiation
- OTUs depend on the data set: no comparability of data sets
Go Up
ASV Denoising
Amplicon sequence variant (ASV) denoising is a method used to remove errors from amplicon sequencing data.
The goal of ASV denoising is to identify true biological sequences and remove errors introduced during
PCR amplification and sequencing.
ASV denoising is a newer method than OTU clustering and is based on a different algorithm based on
a statistical model to estimate the error rate of each nucleotide position in the sequence,
which is then used to correct errors in the data.
- Discriminate sequence variations from sequencing errors
- Based on an error model and the assumption that sequences containing errors are less likely observed repeatedly
Go Up
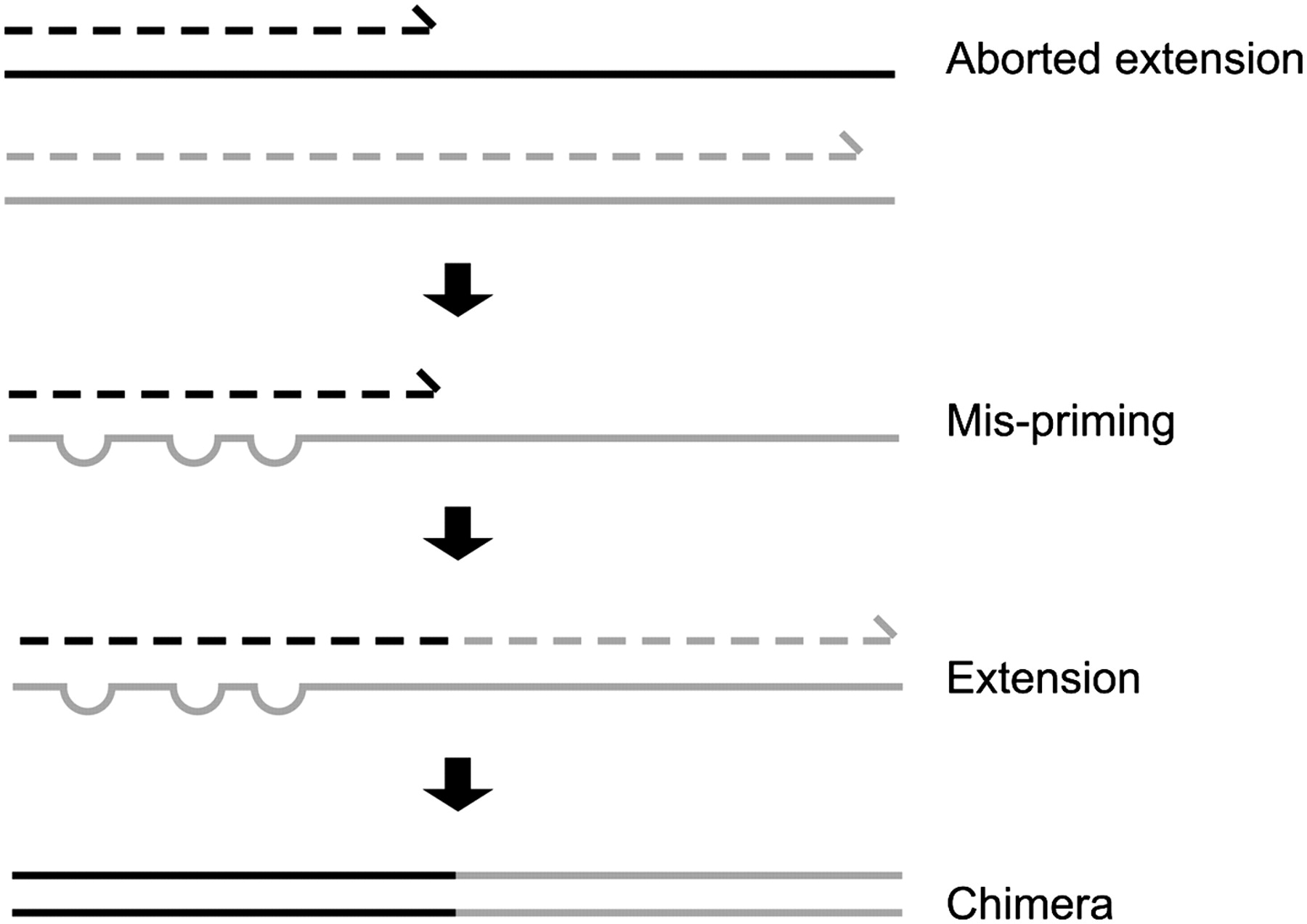
Chimera Removal
Chimera removal is a process used to identify and remove chimeric sequences from a dataset.
Chimeras are sequences that are formed by two or more biological sequences incorrectly joined together.
This often occurs during PCR reactions.
The goal of chimera removal is to identify and remove these sequences so that they do not interfere with downstream analysis.
- During PCR multiple sequences can combine to form a hybrid
- Must be removed from your data for better results
Go Up
Search marker database and taxonomy assignment
A marker database is a database of genes or proteins that are used to identify organisms in a sample.
Taxonomy assignment is the process of assigning taxonomic labels to metagenomic sequences based
on their similarity to sequences in reference databases.
This process is essential for understanding the composition of microbial communities in environmental samples.
- Homology with reference databases
- Amplicon

- Shotgun: MetaPhlAn2 database
- ~1M unique clade-specific marker genes
- ~17,000 reference genomes (bacterial and archaeal, viral and eukaryotic)
- Accuracy depends on quality and completeness of database used
- Databases are inevitable incomplete
Go Up
Search functional database
Functional databases are databases that contain information about the functions of genes or proteins.
These databases can be used to identify the functions of genes or proteins in a sample.
Functional analysis is a main component of metagenomic analysis and is used to figure out what
the organisms in a sample are actually doing.
One common approach is to query a specific set of known sequences that have an associated function,
or can be related to a series of function.
- Databases to identify gene families
- Grouping in other functional categories
- MetaCyc Reactions
- KEGG Orthogroups (KOs)
- Pfam domains
- Level-4 enzyme commission (EC) categories
- EggNOG (including COGs)
- Gene Ontology (GO)
- Informative GO
- Pathway reconstruction
Go Up
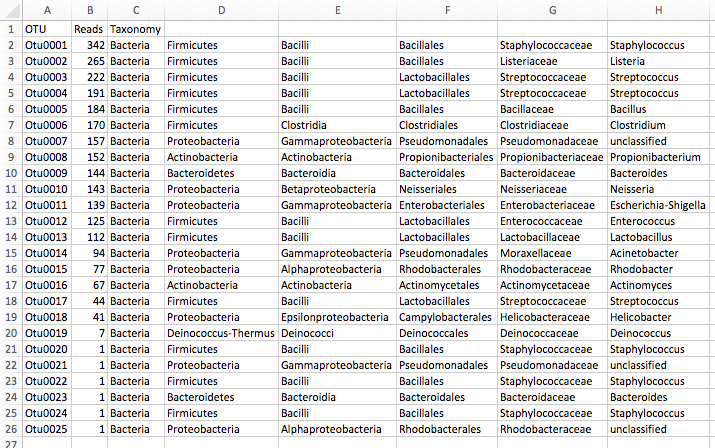
Results: OTU table
Go Up
Results: Visualizations
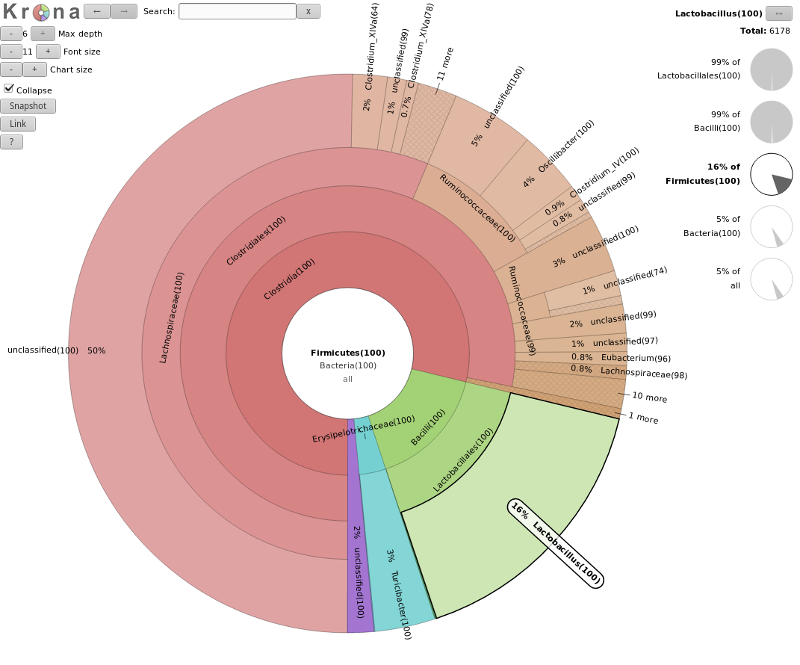
Krona
- Interactive exploration of sample taxonomy
Go Up
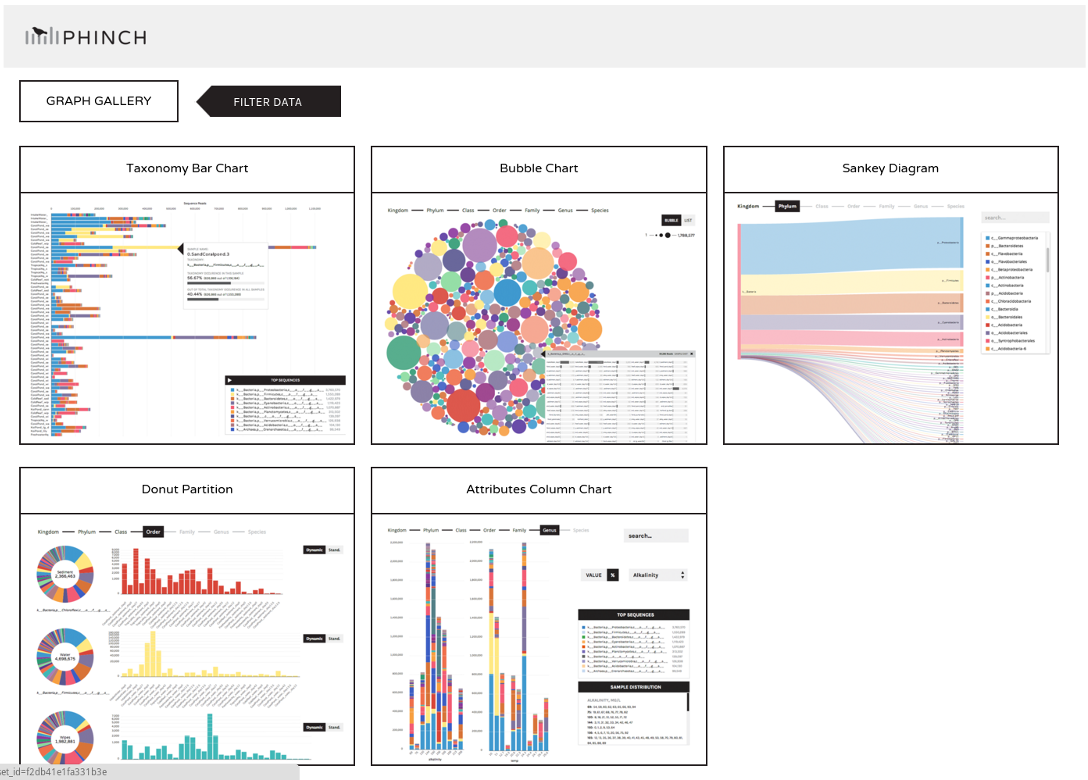
Phinch
- BIOM file input
- various visualizations
- multi-sample data
Go Up