What is the effect of normal variation in the gut microbiome on host health?
In this tutorial we will perform an analysis based on the Standard Operating Procedure (SOP) for MiSeq data, developed by the Schloss lab, the creators of the mothur software package.
NOTE: In this tutorial we use 16S rRNA data, but similar pipelines can be used for WGS data.
In this tutorial we use the dataset generated by the Schloss lab to illustrate their MiSeq SOP.
They describe the experiment as follows:
“The Schloss lab is interested in understanding the effect of normal variation in the gut microbiome on host health. To that end, we collected fresh feces from mice on a daily basis for 365 days post weaning. During the first 150 days post weaning (dpw), nothing was done to our mice except allow them to eat, get fat, and be merry.
We were curious whether the rapid change in weight observed during the first 10 dpw affected the stability microbiome compared to the microbiome observed between days 140 and 150.”
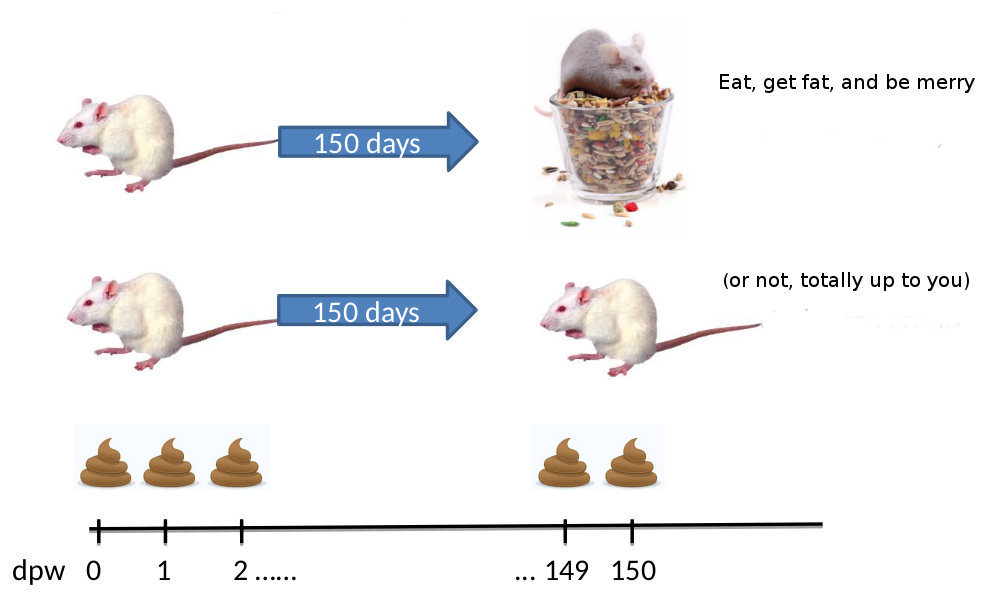
To speed up analysis for this tutorial, we will use only a subset of this data. We will look at a single mouse at 10 different time points (5 early, 5 late).
In order to assess the error rate of the analysis pipeline and experimental setup,
the Schloss lab additionally sequenced a mock community with a known composition
(genomic DNA from 21 bacterial strains).
The sequences used for this mock sample are contained in the file HMP_MOCK.v35.fasta
For this tutorial, you are given 10 pairs of files. For example, the following pair of files:
F3D0_S188_L001_R1_001.fastq
F3D0_S188_L001_R2_001.fastq
The first part of the file name indicates the sample; F3D0 here signifies that this sample was obtained from Female 3 on Day 0.
The rest of the file name is identical, except for _R1 and _R2, this is used to indicate the forward and reverse reads respectively.
https://zenodo.org/record/800651/files/F3D0_R1.fastq
https://zenodo.org/record/800651/files/F3D0_R2.fastq
https://zenodo.org/record/800651/files/F3D141_R1.fastq
https://zenodo.org/record/800651/files/F3D141_R2.fastq
https://zenodo.org/record/800651/files/F3D142_R1.fastq
https://zenodo.org/record/800651/files/F3D142_R2.fastq
https://zenodo.org/record/800651/files/F3D143_R1.fastq
https://zenodo.org/record/800651/files/F3D143_R2.fastq
https://zenodo.org/record/800651/files/F3D144_R1.fastq
https://zenodo.org/record/800651/files/F3D144_R2.fastq
https://zenodo.org/record/800651/files/F3D145_R1.fastq
https://zenodo.org/record/800651/files/F3D145_R2.fastq
https://zenodo.org/record/800651/files/F3D146_R1.fastq
https://zenodo.org/record/800651/files/F3D146_R2.fastq
https://zenodo.org/record/800651/files/F3D147_R1.fastq
https://zenodo.org/record/800651/files/F3D147_R2.fastq
https://zenodo.org/record/800651/files/F3D148_R1.fastq
https://zenodo.org/record/800651/files/F3D148_R2.fastq
https://zenodo.org/record/800651/files/F3D149_R1.fastq
https://zenodo.org/record/800651/files/F3D149_R2.fastq
https://zenodo.org/record/800651/files/F3D150_R1.fastq
https://zenodo.org/record/800651/files/F3D150_R2.fastq
https://zenodo.org/record/800651/files/F3D1_R1.fastq
https://zenodo.org/record/800651/files/F3D1_R2.fastq
https://zenodo.org/record/800651/files/F3D2_R1.fastq
https://zenodo.org/record/800651/files/F3D2_R2.fastq
https://zenodo.org/record/800651/files/F3D3_R1.fastq
https://zenodo.org/record/800651/files/F3D3_R2.fastq
https://zenodo.org/record/800651/files/F3D5_R1.fastq
https://zenodo.org/record/800651/files/F3D5_R2.fastq
https://zenodo.org/record/800651/files/F3D6_R1.fastq
https://zenodo.org/record/800651/files/F3D6_R2.fastq
https://zenodo.org/record/800651/files/F3D7_R1.fastq
https://zenodo.org/record/800651/files/F3D7_R2.fastq
https://zenodo.org/record/800651/files/F3D8_R1.fastq
https://zenodo.org/record/800651/files/F3D8_R2.fastq
https://zenodo.org/record/800651/files/F3D9_R1.fastq
https://zenodo.org/record/800651/files/F3D9_R2.fastq
https://zenodo.org/record/800651/files/Mock_R1.fastq
https://zenodo.org/record/800651/files/Mock_R2.fastq
https://zenodo.org/record/800651/files/HMP_MOCK.v35.fasta
https://zenodo.org/record/800651/files/silva.v4.fasta
https://zenodo.org/record/800651/files/trainset9_032012.pds.fasta
https://zenodo.org/record/800651/files/trainset9_032012.pds.tax
https://zenodo.org/record/800651/files/mouse.dpw.metadata
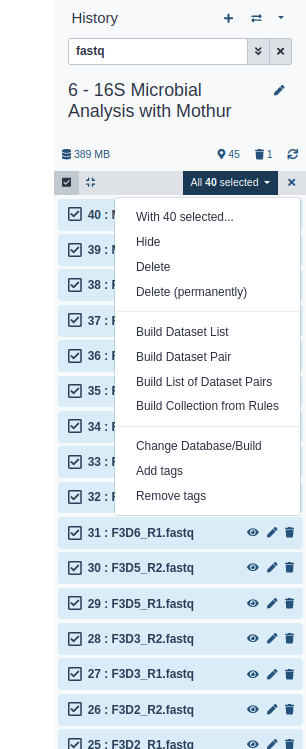
Now that’s a lot of files to manage. Luckily Galaxy can make life a bit easier by allowing us to create dataset collections. This enables us to easily run tools on multiple datasets at once.
Since we have paired-end data, each sample consist of two separate fastq files, one containing the forward reads, and one containing the reverse reads.
We can recognize the pairing from the file names, which will differ only by _R1 or _R2 in the filename.
We can tell Galaxy about this paired naming convention, so that our tools will know which files belong together.
We do this by building a List of Dataset Pairs
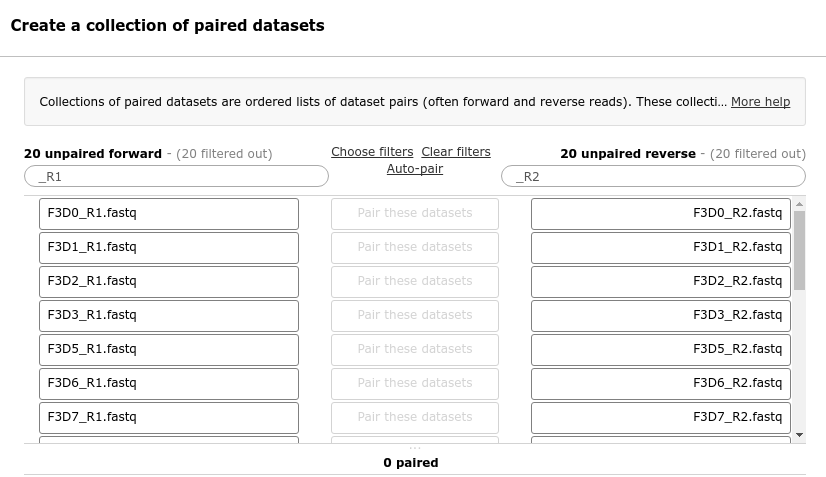
In the next dialog window you can create the list of pairs. By default Galaxy will look for pairs of files that differ only by a _1 and _2 part in their names. In our case however, these should be _R1 and _R2.
Forward: _R1, Reverse: _R2 (note that you can also enter Filters manually in the text fields on the top)
You should now see a list of pairs suggested by Galaxy:
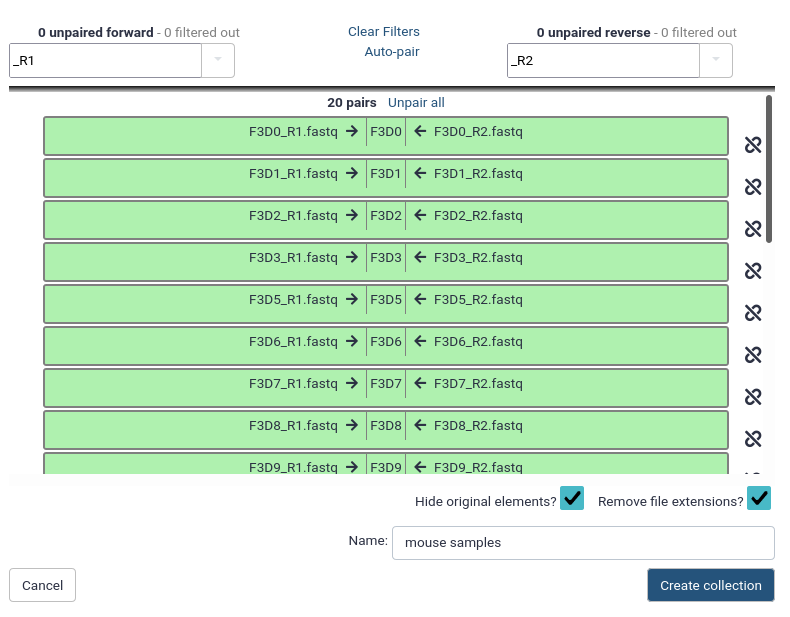
Before starting any analysis, it is always a good idea to assess the quality of your input data and improve it where possible by trimming and filtering reads.
The mothur toolsuite contains several tools to assist with this task.
In this experiment, paired-end sequencing of the ~253 bp V4 region of the 16S rRNA gene was performed. The sequencing was done from either end of each fragment. Because the reads are about 250 bp in length, this results in a significant overlap between the forward and reverse reads in each pair. We will combine these pairs of reads into contigs.
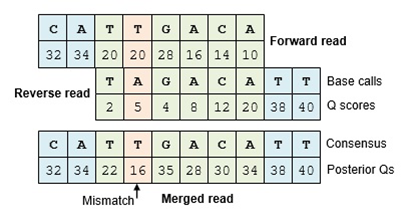
The Make.contigs tool creates the contigs, and uses the paired collection as input:
Where an overlapping base call differs between the two reads, the quality score is used to determine the consensus base call.
A new quality score is derived by combining the two original quality scores in both of the reads for all the overlapping positions.`
Make.contigs Tool with the following parameters:
This step combined the forward and reverse reads for each sample, and also combined the resulting contigs from all samples into a single file.
So we have gone from a paired collection of 20x2 FASTQ files, to a single FASTA file.
In order to retain information about which reads originated from which samples, the tool also output a group file. View that file now, it should look something like this:
As the next step, we want to improve the quality of our data. But first, let’s get a feel of our dataset:
Summary.seqs Tool with the following parameters:
Make.contigs toolThe summary output files give information per read. The logfile outputs also contain some summary statistics:
Start End NBases Ambigs Polymer NumSeqs
Minimum: 1 248 248 0 3 1
2.5%-tile: 1 252 252 0 3 3810
25%-tile: 1 252 252 0 4 38091
Median: 1 252 252 0 4 76181
75%-tile: 1 253 253 0 5 114271
97.5%-tile: 1 253 253 6 6 148552
Maximum: 1 502 502 249 243 152360
Mean: 1 252.811 252.811 0.70063 4.44854
# of Seqs: 152360
In this dataset:
Our region of interest, the V4 region of the 16S gene, is only around 250 bases long.
Any reads significantly longer than this expected value likely did not assemble well in the Make.contigs step.
Furthermore, we see that 2,5% of our reads had between 6 and 249 ambiguous base calls (Ambigs column). In the next steps we will clean up our data by removing these problematic reads.
We do this data cleaning using the Screen.seqs tool, which removes:
Screen.seqs Tool with the following parameters
Make.contigs toolMake.contigs tool stepQuestion How many reads were removed in this screening step?
Because we are sequencing many of the same organisms, we anticipate that many of our sequences are duplicates of each other.
Unique.seqs tool with the following parameters:
Screen.seqsView the resulting datasets.
Unique.seqs tool outputs two files:
Question
Here we see that this step has greatly reduced the size of our sequence file; not only will this speed up further computational steps, it will also greatly reduce the amount of disk space (and your Galaxy quota) needed to store all the intermediate files generated during this analysis.
To recap, we now have the following files:
Unique.seqsUnique.seqs)Screen.seqs)To further reduce file sizes and streamline analysis, we can use the Count.seqs tool to combine the group file and the names file into a single count table.
Count.seqs tool with the following parameters:
Unique.seqsScreen.seqsView the resulting datasets.
The first column contains the read names of the representative sequences, and the subsequent columns contain the number of duplicates of this sequence observed in each sample.
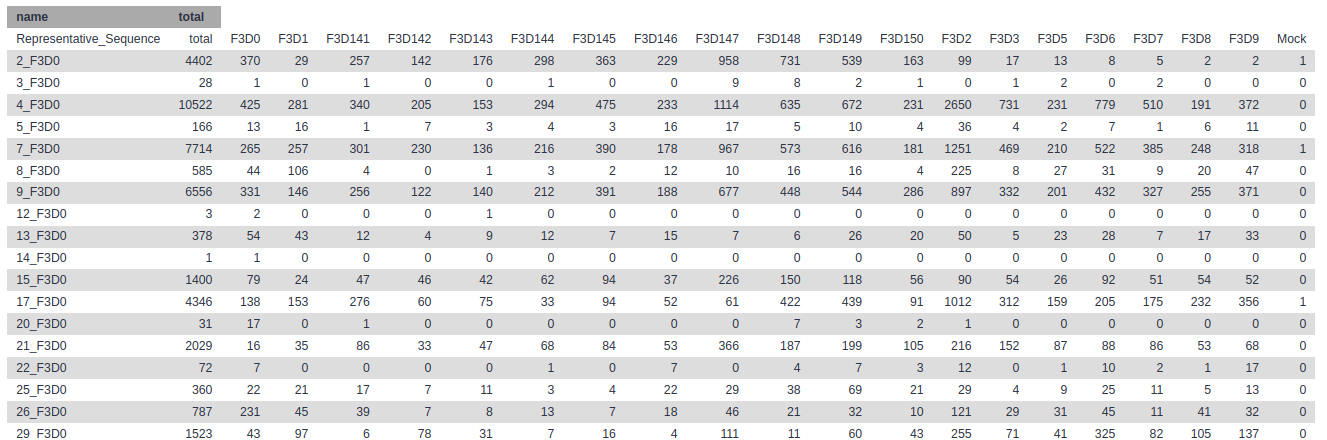
We are now ready to align our sequences to the reference alignment. This is an important step to improve the clustering of your OTUs.
In mothur this is done by determining for each unique sequence the entry of the reference database
that has the most k-mers in common (i.e. the most substring of fixed length k).
For the reference sequence with the most common k-mers and the unique sequence a standard global sequence alignment is computed (using the Needleman-Wunsch algorithm).
Align.seqs Tool with the following parameters:
Unique.seqs toolAt first glance, it might look like there is not much information there. We see our read names, but only period . characters below it.
>2_F3D0
............................................................................
>3_F3D0
............................................................................
>4_F3D0
............................................................................
This is because the V4 region is located further down our reference database and nothing aligns to the start of it.
If you scroll to right you will start seeing some more informative bits:
.....T-------AC---GG-AG-GAT------------
Here we start seeing how our sequences align to the reference database. There are different alignment characters in this output:
.: terminal gap character (before the first or after the last base in our query sequence)-: gap character within the query sequenceWe will cut out only the V4 region in a later step (Filter.seqs tool)
Summary.seqs Tool with the following parameters:
Align.seqs toolCount.seqs toolView the log output.
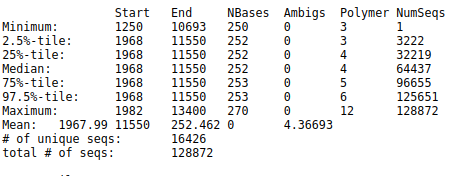
The Start and End columns tell us that the majority of reads aligned between positions 1968 and 11550,
which is what we expect to find given the reference file we used.
However, some reads align to very different positions, which could indicate insertions or deletions at the terminal ends of the alignments or other complicating factors.
Also notice the Polymer column in the output table.
This indicates the average homopolymer length.
Since we know that our reference database does not contain any homopolymer stretches longer than 8 reads,
any reads containing such long stretches are likely the result of PCR errors and we would be wise to remove them.
Next we will clean our data further by removing poorly aligned sequences and any sequences with long homopolymer stretches.
To ensure that all our reads overlap our region of interest, we will:
Screen.seqs tool.Filter.seqs tool.-, or . for terminal gaps) at that position in every sequence (also using Filter.seqs tool).Unique.seqs once more.Pre.cluster tool. Sequences that only differ by one or two bases at this point are likely to represent sequencing errors rather than true biological variation, so we will cluster such sequences together.Screen.seqs Tool with the following parameters:
Align.seqs toolCount.seqsFilter.seqs Tool with the following parameters:
Screen.seqs tool>2_F3D0
TAC--GG-AG-GAT--GCG-A-G-C-G-T-T--AT-C-CGG-AT--TT-A-T-T--GG-GT--TT-A-AA-GG-GT-GC-G-TA-GGC-G-G-C-CT-G-C-C-AA-G-T-C-A-G-C-G-G--TA-A-AA-TT-G-C-GG-GG--CT-C-AA-C-C-C-C-G-T-A--CA-G-C-CGTT-GAAAC-TG-C-CGGGC-TCGA-GT-GG-GC-GA-G-A---AG-T-A-TGCGGAATGCGTGGTGT-AGCGGT-GAAATGCATAG-AT-A-TC-AC-GC-AG-AACCCCGAT-TGCGAAGGCA------GC-ATA-CCG-G-CG-CC-C-T-ACTGACG-CTGA-GGCA-CGAAA-GTG-CGGGG-ATC-AAACAGG
>3_F3D0
TAC--GG-AG-GAT--GCG-A-G-C-G-T-T--GT-C-CGG-AA--TC-A-C-T--GG-GC--GT-A-AA-GG-GC-GC-G-TA-GGC-G-G-T-TT-A-A-T-AA-G-T-C-A-G-T-G-G--TG-A-AA-AC-T-G-AG-GG--CT-C-AA-C-C-C-T-C-A-G-CCT-G-C-CACT-GATAC-TG-T-TAGAC-TTGA-GT-AT-GG-AA-G-A---GG-A-G-AATGGAATTCCTAGTGT-AGCGGT-GAAATGCGTAG-AT-A-TT-AG-GA-GG-AACACCAGT-GGCGAAGGCG------AT-TCT-CTG-G-GC-CA-A-G-ACTGACG-CTGA-GGCG-CGAAA-GCG-TGGGG-AGC-AAACAGG
>4_F3D0
TAC--GG-AG-GAT--GCG-A-G-C-G-T-T--AT-C-CGG-AT--TT-A-T-T--GG-GT--TT-A-AA-GG-GT-GC-G-CA-GGC-G-G-A-AG-A-T-C-AA-G-T-C-A-G-C-G-G--TA-A-AA-TT-G-A-GA-GG--CT-C-AA-C-C-T-C-T-T-C--GA-G-C-CGTT-GAAAC-TG-G-TTTTC-TTGA-GT-GA-GC-GA-G-A---AG-T-A-TGCGGAATGCGTGGTGT-AGCGGT-GAAATGCATAG-AT-A-TC-AC-GC-AG-AACTCCGAT-TGCGAAGGCA------GC-ATA-CCG-G-CG-CT-C-A-ACTGACG-CTCA-TGCA-CGAAA-GTG-TGGGT-ATC-GAACAGG
>5_F3D0
TAC--GT-AG-GTG--GCA-A-G-C-G-T-T--AT-C-CGG-AT--TT-A-C-T--GG-GT--GT-A-AA-GG-GC-GT-G-TA-GGC-G-G-G-GA-C-G-C-AA-G-T-C-A-G-A-T-G--TG-A-AA-AC-C-A-CG-GG--CT-C-AA-C-C-T-G-T-G-G-CCT-G-C-ATTT-GAAAC-TG-T-GTTTC-TTGA-GT-AC-TG-GA-G-A---GG-C-A-GACGGAATTCCTAGTGT-AGCGGT-GAAATGCGTAG-AT-A-TT-AG-GA-GG-AACACCAGT-GGCGAAGGCG------GT-CTG-CTG-G-AC-AG-C-A-ACTGACG-CTGA-GGCG-CGAAA-GCG-TGGGG-AGC-AAACAGG
These are all our representative reads again, now with additional alignment information.
View the log output
Length of filtered alignment: 376
Number of columns removed: 13049
Length of the original alignment: 13425
Number of sequences used to construct filter: 16298
From this log file we see that while our initial alignment was 13425 positions wide,
after filtering the overhangs (trump parameter) and removing positions that had a gap in every aligned read
(vertical parameter), we have trimmed our alignment down to a length of 376.
Because any filtering step we perform might lead to sequences no longer being unique, we deduplicate our data
Unique.seqs Tool with the following parameters:
Filter.seqs toolScreen.seqsQuestion
The next step in cleaning our data, is to merge near-identical sequences together. Sequences that only differ by around 1 in every 100 bases are likely to represent sequencing errors, not true biological variation.
Because our contigs are ~250 bp long, we will set the threshold to 2 mismatches.
Pre.cluster Tool with the following parameters:
Unique.seqs tool runUnique.seqs toolQuestion
We have now thoroughly cleaned our data and removed as much sequencing error as we can. Next, we will look at a class of sequencing artefacts known as chimeras.

During PCR amplification, it is possible that two unrelated templates are combined to form a sort of hybrid sequence,
also called a chimera. Needless to say, we do not want such sequencing artefacts confounding our results.
We’ll do this chimera removal using the VSEARCH algorithm that is called within mothur, using the Chimera.vsearch tool.
Chimera.vsearch Tool with the following parameters:
Pre.cluster toolPre.cluster toolRunning Chimera.vsearch with the count file will remove the chimeric sequences from the count table, but we still need to remove those sequences from the fasta file as well.
Remove.seqs Tool with the following parameters:
Chimera.vsearch toolPre.cluster toolChimera.vsearchQuestion
Looking at the chimera.vsearch accnos output, we see that 3,439 representative sequences were flagged as chimeric.
If we run summary.seqs on the resulting fasta file and count table,
we see that we went from 128,655 sequences down to 118,091 total sequences in this step,
for a reduction of 10,564 total sequences, or 8.2%.
This is a reasonable number of sequences to be flagged as chimeric.
Now that we have thoroughly cleaned our data, we are finally ready to assign a taxonomy to our sequences.
We will do this using a Bayesian classifier (via the Classify.seqs tool) and a mothur-formatted training set
provided by the Schloss lab based on the
RDP (Ribosomal Database Project, Cole et al. 2013) reference taxonomy.
NOTE! In this tutorial we will use the RDP classifier and reference taxonomy for classification, but there are several different taxonomic assignment algorithms and reference databases available for this purpose.
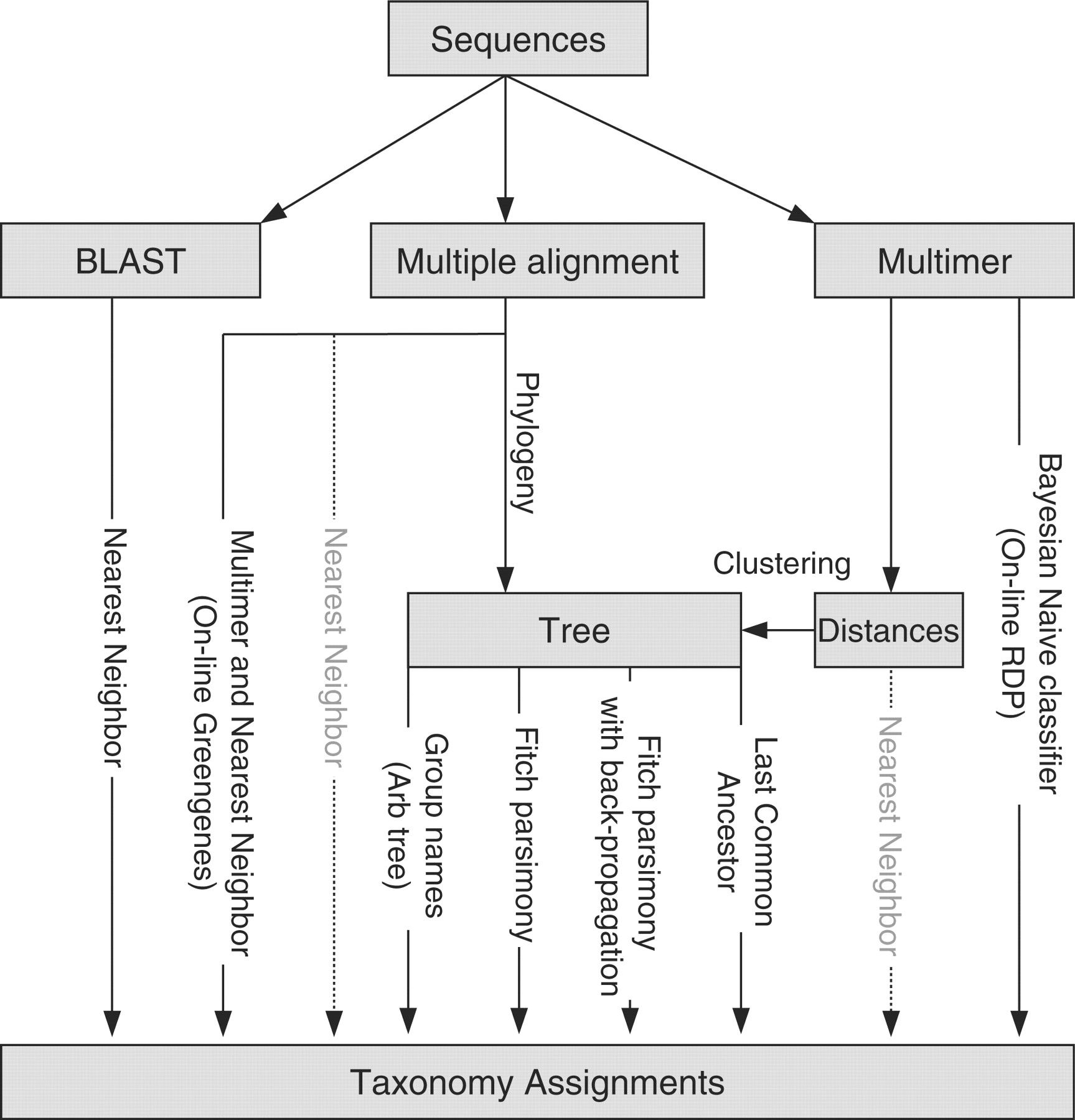
The most commonly used reference taxonomies are:
Which reference taxonomy is best for your experiments depends on a number of factors such as the type of sample and variable region sequenced.
Another discussion about how these different databases compare was described by Balvočiūtė and Huson 2017.
Despite all we have done to improve data quality, there may still be more to do: there may be 18S rRNA gene fragments or 16S rRNA from Archaea, chloroplasts, and mitochondria that have survived all the cleaning steps up to this point.
We are generally not interested in these sequences and want to remove them from our dataset
Classify.seqs Tool with the following parameters
Remove.seqs toolRemove.seqsRemove.lineage Tool with the following parameters
Classify.seqs toolRemove.seqs toolRemove.seqsQuestion
The data is now as clean as we can get it.
This step is only possible if you have co-sequenced a mock community with your samples. A mock community is a sample of which you know the exact composition and is something we recommend to do, because it will give you an idea of how accurate your sequencing and analysis protocol is.
What is a mock community?
A mock community is an artificially constructed sample; a defined mixture of microbial cells and/or viruses or nucleic acid molecules created in vitro to simulate the composition of a microbiome sample or the nucleic acid isolated therefrom.
Why sequence a mock community?
In a mock community, we know exactly which sequences/organisms we expect to find, and at which proportions.
Therefore, we can use such an artificial sample to assess the error rates of our sequencing and analysis pipeline.
If our workflow performed well on the mock sample, we have more confidence in the accuracy of the results on the rest of our samples.
The mock community in this experiment was composed of genomic DNA from 21 bacterial strains. So in a perfect world, this is exactly what we would expect the analysis to produce as a result.
Get.groups Tool with the following parameters
Remove.lineage toolRemove.lineage tool Selected 58 sequences from your fasta file.
Selected 4046 sequences from your count file.
The Mock sample has 58 unique sequences, representing a total of 4,046 total sequences.
The Seq.error tool measures the error rates using our mock reference.
Here we align the reads from our mock sample back to their known sequences, to see how many fail to match.
Seq.error Tool with the following parameters
Get.groups toolGet.groups toolOverall error rate: 6.5108e-05
Errors Sequences
0 3998
1 3
2 0
3 2
4 1
[..]
That is pretty good! The error rate is only 0.0065%! This gives us confidence that the rest of the samples are also of high quality, and we can continue with our analysis.
We will now estimate the accuracy of our sequencing and analysis pipeline by clustering the Mock sequences into OTUs, and comparing the results with the expected outcome.
For this a distance matrix is calculated (i.e. the distances between all pairs of sequences). From this distance matrix a clustering is derived using the OptiClust algorithm:
Let’s Run
Dist.seqs Tool the following parameters
Get.groups toolCluster - Assign sequences to OTUs Tool with the following parameters
Dist.seqs toolGet.groups toolNow we make a shared file that summarizes all our data into one handy table
Make.shared Tool with the following parameters
Cluster - Assign sequences to OTUs toolGet.groups toolAnd now we generate intra-sample rarefaction curves
Rarefaction.single Tool with the following parameters
Make.sharedQuestion
Open the rarefaction output (dataset named sobs inside the rarefaction curves output collection), it should look something like this:
numsampled 0.03- lci- hci-
1 1.0000 1.0000 1.0000
100 18.1230 16.0000 20.0000
200 19.2930 18.0000 21.0000
300 19.9460 18.0000 22.0000
400 20.4630 19.0000 23.0000
500 20.9330 19.0000 24.0000
600 21.3830 19.0000 24.0000
700 21.8280 19.0000 25.0000
800 22.2560 20.0000 26.0000
900 22.6520 20.0000 26.0000
1000 23.0500 20.0000 27.0000
1100 23.4790 20.0000 27.0000
1200 23.8600 21.0000 28.0000
1300 24.2400 21.0000 28.0000
1400 24.6210 21.0000 28.0000
1500 24.9980 22.0000 29.0000
1600 25.3890 22.0000 29.0000
1700 25.7870 22.0000 30.0000
1800 26.1640 23.0000 30.0000
1900 26.5590 23.0000 30.0000
2000 26.9490 23.0000 31.0000
2100 27.3170 24.0000 31.0000
2200 27.6720 24.0000 31.0000
2300 28.0110 24.0000 32.0000
2400 28.3530 25.0000 32.0000
2500 28.7230 25.0000 32.0000
2600 29.1040 26.0000 32.0000
2700 29.4250 26.0000 33.0000
2800 29.7860 27.0000 33.0000
2900 30.1420 27.0000 33.0000
3000 30.4970 27.0000 33.0000
3100 30.8070 28.0000 33.0000
3200 31.1450 28.0000 34.0000
3300 31.4990 28.0000 34.0000
3400 31.8520 29.0000 34.0000
3500 32.1970 29.0000 34.0000
3600 32.5280 30.0000 34.0000
3700 32.8290 30.0000 34.0000
3800 33.1710 31.0000 34.0000
3900 33.5000 32.0000 34.0000
4000 33.8530 33.0000 34.0000
4046 34.0000 34.0000 34.0000
When we use the full set of 4060 sequences, we find 34 OTUs from the Mock community; and with 3000 sequences, we find about 31 OTUs.
In an ideal world, we would find exactly 21 OTUs. Despite our best efforts, some chimeras or other contaminations may have slipped through our filtering steps.
Now that we have assessed our error rates we are ready for some real analysis.
Now that we have cleaned up our data set as best we can, and assured ourselves of the quality of our sequencing pipeline by considering a mock sample, we are almost ready to cluster and classify our real data.
But before we start, we should first remove the Mock dataset from our data, as we no longer need it.
Remove.groups Tool with the following parameters
Remove.lineage toolRemove.lineage toolRemove.lineageThere are several ways we can perform clustering.
For the Mock community, we used the traditional approach of using the Dist.seqs and Cluster tools.
Alternatively, we can also use the Cluster.split tool.
With this approach, the sequences are split into bins, and then clustered with each bin.
Taxonomic information is used to guide this process.
The Schloss lab have published results showing that if you split at the level of Order or Family, and cluster to a 0.03 cutoff, you’ll get just as good of clustering as you would with the “traditional” approach.
In addition, this approach is less computationally expensive and can be parallelized, which is especially advantageous when you have large datasets.
Cluster.split Tool with the following parameters
Remove.groups toolRemove.groups toolRemove.groups toolNext we want to know how many sequences are in each OTU from each group and we can do this using
the Make.shared tool. Here we tell mothur that we’re really only interested in the 0.03 cutoff level
Make.shared Tool with the following parameters
Cluster.split toolRemove.groups toolWe probably also want to know the taxonomy for each of our OTUs.
Classify.otu Tool with the following parameters
Cluster.split toolRemove.groups toolRemove.groups toolOTU Size Taxonomy
...
Otu008 5260 Bacteria(100);"Bacteroidetes"(100);"Bacteroidia"(100);"Bacteroidales"(100);"Rikenellaceae"(100);Alistipes(100);
Otu009 3605 Bacteria(100);"Bacteroidetes"(100);"Bacteroidia"(100);"Bacteroidales"(100);"Porphyromonadaceae"(100);"Porphyromonadaceae"_unclassified(100);
Otu010 3058 Bacteria(100);Firmicutes(100);Bacilli(100);Lactobacillales(100);Lactobacillaceae(100);Lactobacillus(100);
Otu011 2935 Bacteria(100);"Bacteroidetes"(100);"Bacteroidia"(100);"Bacteroidales"(100);"Porphyromonadaceae"(100);"Porphyromonadaceae"_unclassified(100);
Otu012 2078 Bacteria(100);"Bacteroidetes"(100);"Bacteroidia"(100);"Bacteroidales"(100);"Porphyromonadaceae"(100);"Porphyromonadaceae"_unclassified(100);
Otu013 1856 Bacteria(100);Firmicutes(100);Bacilli(100);Lactobacillales(100);Lactobacillaceae(100);Lactobacillus(100);
Otu014 1455 Bacteria(100);Firmicutes(100);Clostridia(100);Clostridiales(100);Lachnospiraceae(100);Lachnospiraceae_unclassified(100);
Otu015 1320 Bacteria(100);Firmicutes(100);Clostridia(100);Clostridiales(100);Lachnospiraceae(100);Lachnospiraceae_unclassified(100);
Otu016 1292 Bacteria(100);Firmicutes(100);Erysipelotrichia(100);Erysipelotrichales(100);Erysipelotrichaceae(100);Turicibacter(100);
The first line shown in the snippet above indicates that Otu008 occurred 5260 times, and that all of the sequences (100%) were binned in the genus Alistipes.
Question
Before we continue, let’s remind ourselves what we set out to do. Our original question was about the stability of the microbiome and whether we could observe any change in community structure between the early and late samples.
Because some of our sample may contain more sequences than others, it is generally a good idea to normalize the dataset by subsampling.
Count.groups Tool with the following parameters
Make.sharedQuestion
Sub.sample Tool with the following parameters
Make.shared toolAll groups (samples) should now have 2389 sequences.
Run count.groups again on the shared output collection by the sub.sample tool to confirm that this is indeed what happened.
Species diversity is a valuable tool for describing the ecological complexity of a single sample (alpha diversity) or between samples (beta diversity).
However, diversity is not a physical quantity that can be measured directly, and many different metrics have been proposed to quantify diversity (Finotello et al. 2016).
Species diversity consists of three components: species richness, taxonomic or phylogenetic diversity and species evenness.
In order to estimate alpha diversity of the samples, we first generate the rarefaction curves.
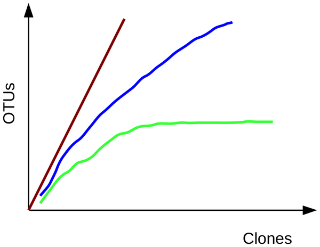
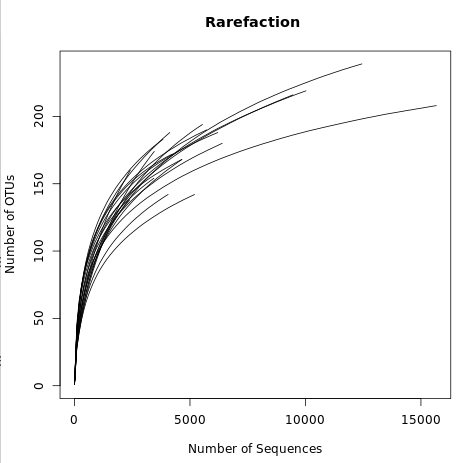
Recall that rarefaction measures the number of observed OTUs as a function of the subsampling size.
A rarefaction curve plots the number of species as a function of the number of individuals sampled. The curve usually begins with a steep slope, which at some point begins to flatten as fewer species are being discovered per sample: the gentler the slope, the less contribution of the sampling to the total number of operational taxonomic units or OTUs.
Rarefaction.single Tool with the following parameters
Make.sharedNote that we used the default diversity measure here (sobs; observed species richness), but there are many more options available under the calc parameter. Descriptions of some of these calculators can be found on the mothur wiki.
Examine the rarefaction curve output.
numsampled 0.03-F3D0 lci-F3D0 hci-F3D0 0.03-F3D1 ...
1 1.0000 1.0000 1.0000 1.0000
100 41.6560 35.0000 48.0000 45.0560
200 59.0330 51.0000 67.0000 61.5740
300 70.5640 62.0000 79.0000 71.4700
400 78.8320 71.0000 87.0000 78.4730
500 85.3650 77.0000 94.0000 83.9990
...
This file displays the number of OTUs identified per amount of sequences used (numsampled). What we would like to see is the number of additional OTUs identified when adding more sequences reaching a plateau.
Then we know we have covered our full diversity. This information would be easier to interpret in the form of a graph.
Plotting tool - for multiple series and graph types Tool with the following parameters
View the rarefaction plot output. From this image can see that the rarefaction curves for all samples have started to level off so we are confident we cover a large part of our sample diversity:
Finally, let’s use the Summary.single tool to generate a summary report. The following step will randomly subsample down to 2389 sequences, repeat this process 1000 times, and report several metrics:
Summary.single Tool with the following parameters
Make.shared toolView the summary output from Summary.single tool.
This shows several alpha diversity metrics:
label group sobs coverage invsimpson invsimpson_lci invsimpson_hci nseqs
0.03 F3D0 167.000000 0.994697 25.686387 24.648040 26.816067 6223.000000
0.03 F3D1 145.000000 0.994030 34.598470 33.062155 36.284520 4690.000000
0.03 F3D141 154.000000 0.991060 19.571632 18.839994 20.362390 4698.000000
0.03 F3D142 141.000000 0.978367 17.029921 16.196090 17.954269 2450.000000
0.03 F3D143 135.000000 0.980738 18.643635 17.593785 19.826728 2440.000000
0.03 F3D144 161.000000 0.980841 15.296728 14.669208 15.980336 3497.000000
0.03 F3D145 169.000000 0.991222 14.927279 14.494740 15.386427 5582.000000
0.03 F3D146 161.000000 0.989167 22.266620 21.201364 23.444586 3877.000000
0.03 F3D147 210.000000 0.995645 15.894802 15.535594 16.271013 12628.000000
0.03 F3D148 176.000000 0.995725 17.788205 17.303206 18.301177 9590.000000
0.03 F3D149 194.000000 0.994957 21.841083 21.280343 22.432174 10114.000000
0.03 F3D150 164.000000 0.989446 23.553161 22.462533 24.755101 4169.000000
0.03 F3D2 179.000000 0.998162 15.186238 14.703161 15.702137 15774.000000
0.03 F3D3 127.000000 0.994167 14.730640 14.180453 15.325243 5315.000000
0.03 F3D5 138.000000 0.990523 29.415378 28.004777 30.975621 3482.000000
0.03 F3D6 155.000000 0.995339 17.732145 17.056822 18.463148 6437.000000
0.03 F3D7 126.000000 0.991916 13.343631 12.831289 13.898588 4082.000000
0.03 F3D8 158.000000 0.992536 23.063894 21.843396 24.428855 4287.000000
0.03 F3D9 162.000000 0.994803 24.120541 23.105499 25.228865 5773.000000
There are a couple of things to note here:
There are many more diversity metrics, and for more information about the different calculators available in mothur, see the mothur wiki page
Beta diversity is a measure of the similarity of the membership and structure found between different samples.
The default calculator in the following section is thetaYC,
which is the Yue & Clayton theta similarity coefficient.
We will also calculate the Jaccard index (termed jclass in mothur).
Dist.shared Tool with the following parameters
Make.shared toolHeatmap.sim Tool with the following parameters
Dist.shared tool (this is a collection input)Look at some of the resulting heatmaps (you may have to download the SVG images first). In all of these heatmaps the red colors indicate communities that are more similar than those with black colors.
For example this is the heatmap for the thetayc calculator (output thetayc.0.03.lt.ave):

and the jclass calulator (output jclass.0.03.lt.ave):

When generating Venn diagrams we are limited by the number of samples that we can analyze simultaneously.
Let’s take a look at the Venn diagrams for the first 4 time points of female 3 using the Venn tool:
Venn Tool with the following parameters

This shows that there were a total of 218 OTUs observed between the 4 time points. Only 73 of those OTUs were shared by all four time points. We could look deeper at the shared file to see whether those OTUs were numerically rare or just had a low incidence.
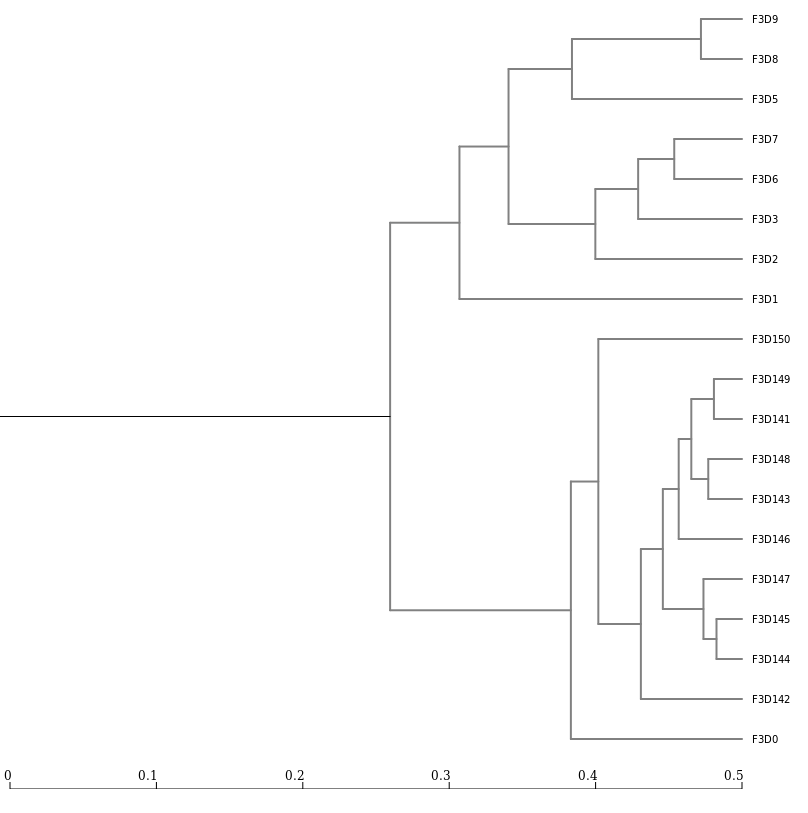
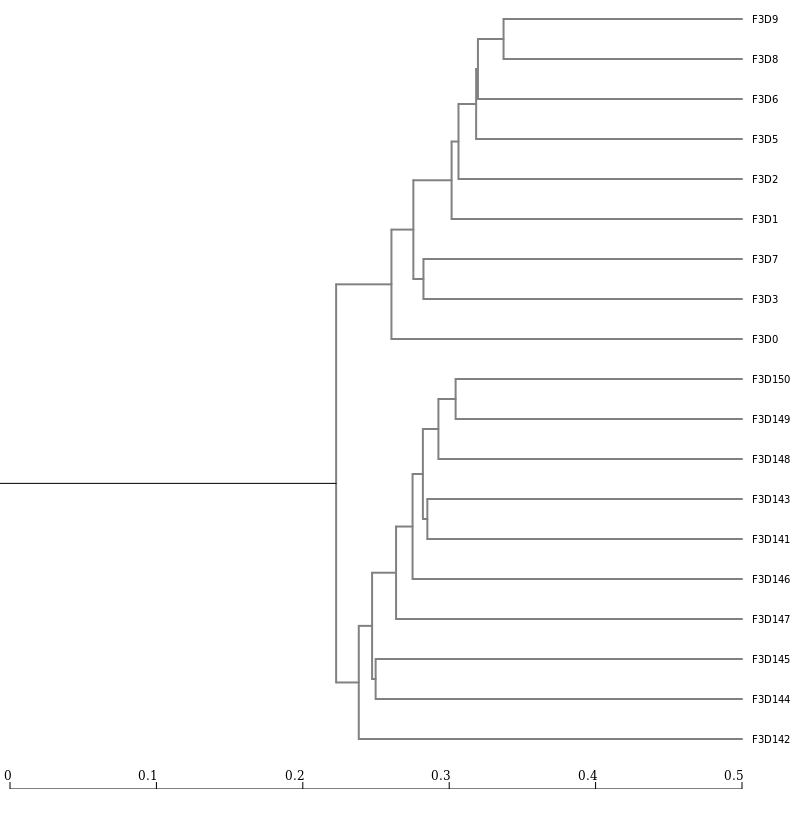
Next, let’s generate a dendrogram to describe the similarity of the samples to each other.
We will generate a dendrogram using the jclass and thetayc calculators within the Tree.shared tool:
Tree.shared Tool with the following parameters
Dist.sharedNewick display Tool with the following parameters
Tree.sharedInspection of the tree shows that the early and late communities cluster with themselves to the exclusion of the others.
thetayc.0.03.lt.ave:
jclass.0.03.lt.ave:
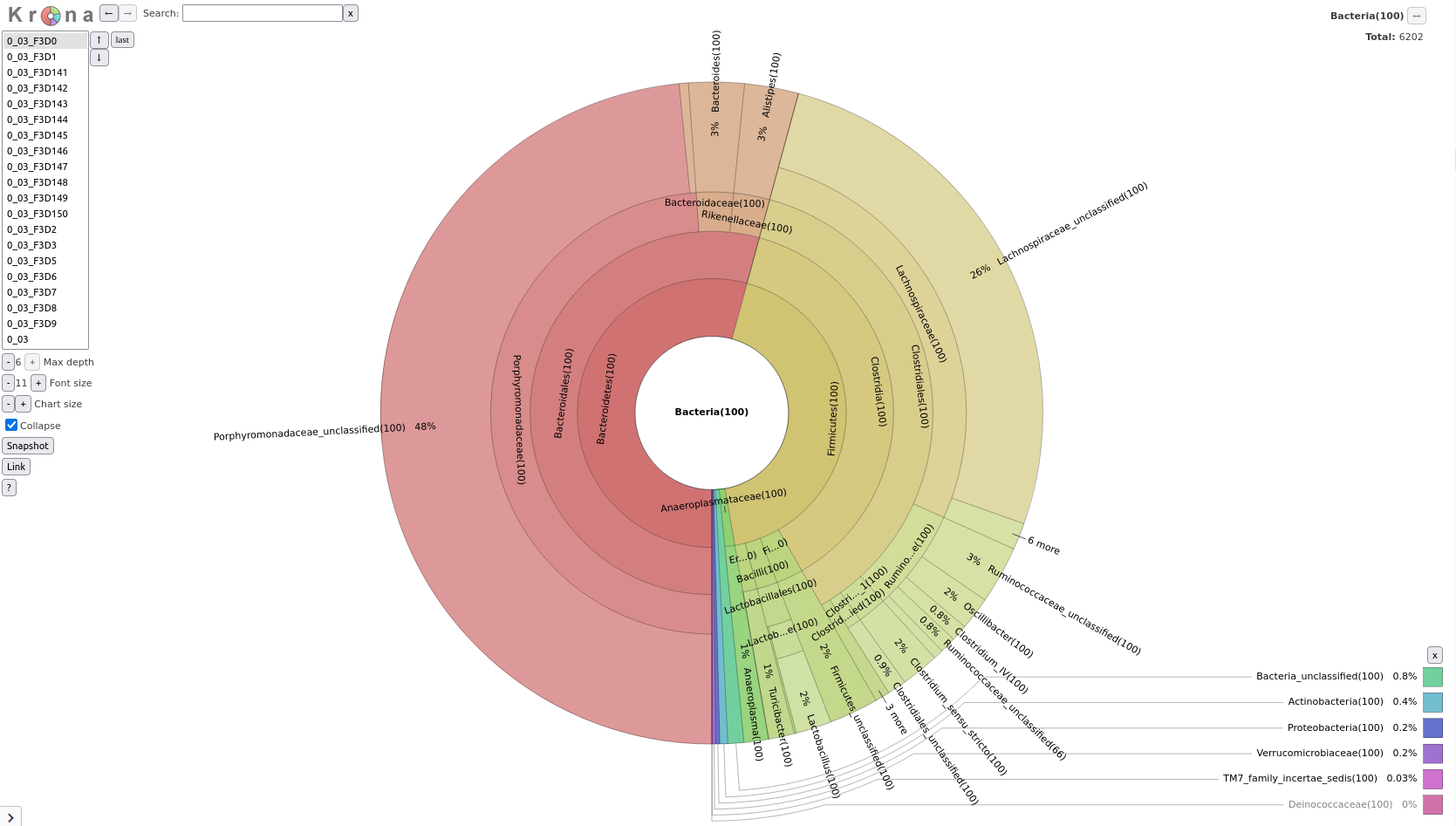
As we saw in the previous lectures, a tool we can use to visualize the composition of our community, is Krona
Taxonomy-to-Krona Tool with the following parameters
Classify.otuKrona pie chart Tool with the following parameters
Taxonomy-to-KronaThe resulting file is an HTML file containing an interactive visualization. For instance try double-clicking the innermost ring labeled “Bacteroidetes”.
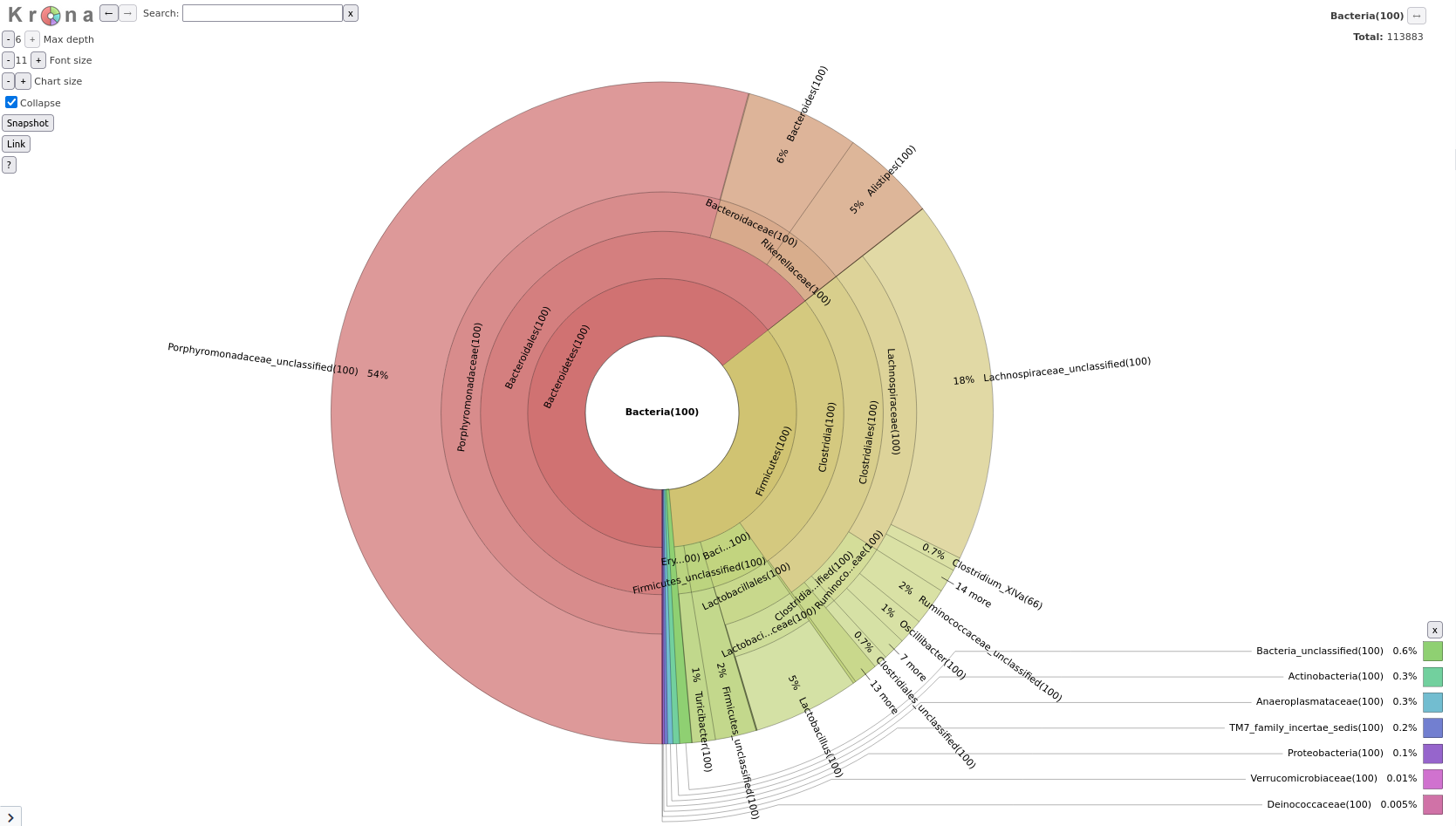
Question
Classify.otu tool we ran earlier
Cluster.split toolRemove.groups toolRemove.groups toolTaxonomy-to-Krona Tool with the following parameters
Classify.otuKrona pie chart Tool with the following parameters
Taxonomy-to-Krona toolThe final result should look something like this (switch between samples via the list on the left):
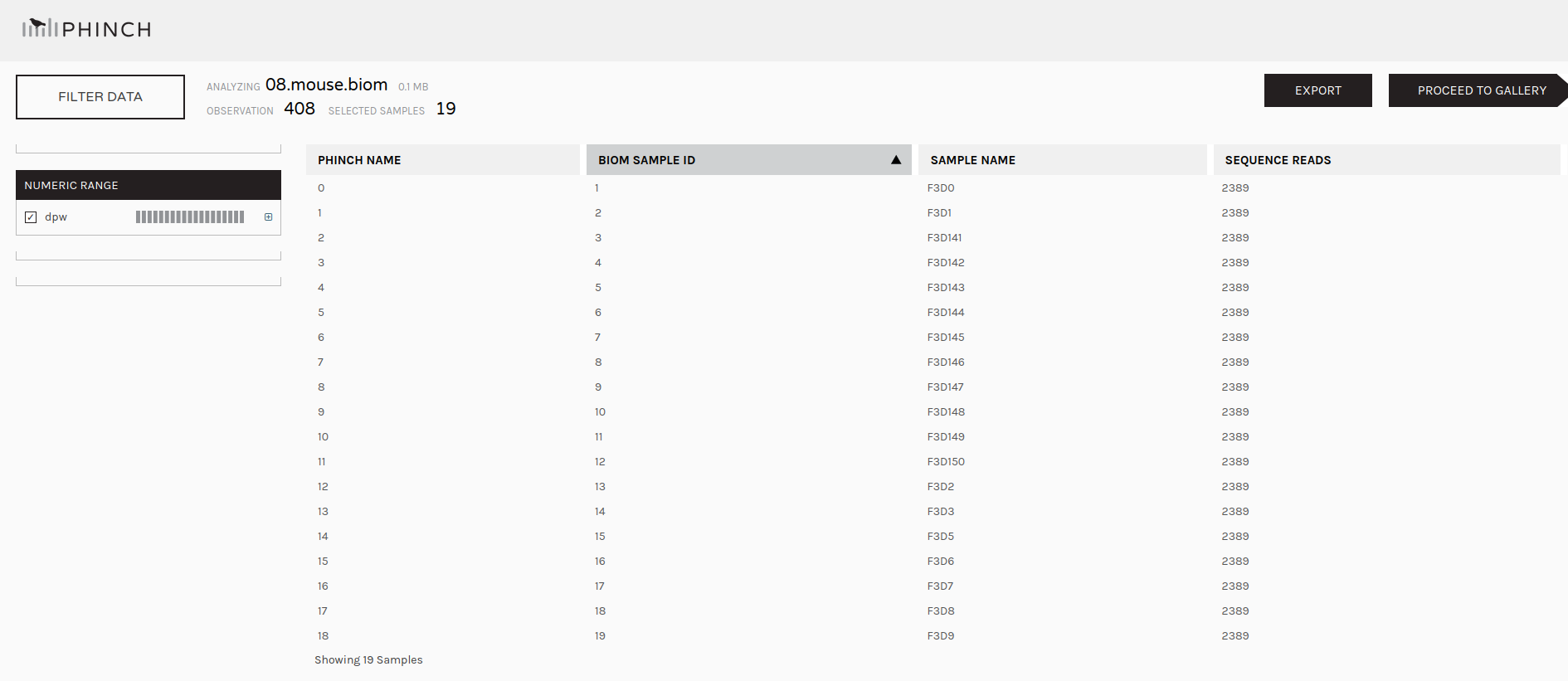
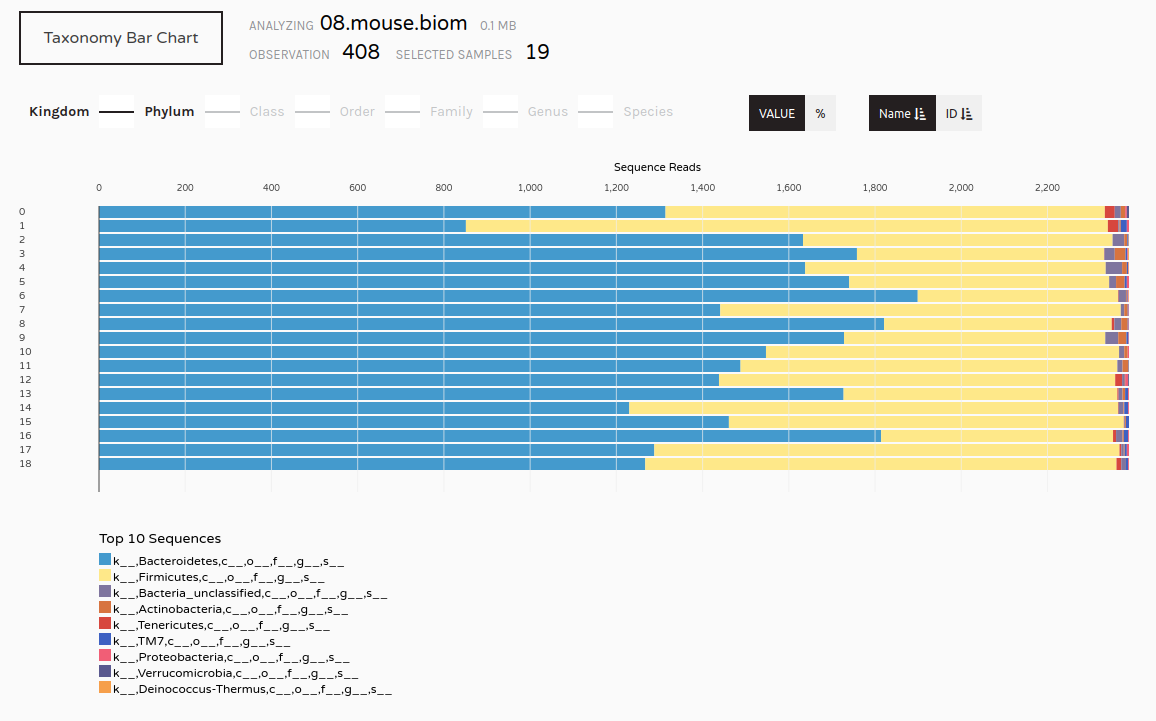
We may now wish to further visualize our results. We can convert our shared file to the more widely used biom format and view it in a platform like Phinch.
Make.biom Tool with the following parameters
Sub.sample tool (collection)Classify.otu toolmouse.dpw.metadata file you uploaded at the start of this tutorialImportant: After downloading, please change the file extension from .biom1 to .biom before uploading to Phinch.