Why metagenomic data should be assembled? What is the difference between co-assembly and individual assembly? What is the difference between reads, contigs and scaffolds? How to assess the quality of metagenomic data assembly?
Metagenomics involves the extraction, sequencing and analysis of combined genomic DNA from entire microbiome samples. It includes then DNA from many different organisms, with different taxonomic background.
Reconstructing the genomes of microorganisms in the sampled communities is critical step in analyzing metagenomic data. To do that, we can use assembly and assemblers, i.e. computational programs that stich together the small fragments of sequenced DNA produced by sequencing instruments.
For a thorough analysis of the community, metagenomic shotgun reads can be assembled with the help of available reference genomes (reference-based assembly) or de novo (de-novo assembly).
Assembling seems intuitively similar to putting together a jigsaw puzzle.
Ideally, the result of a metagenome assembly process should be a set of genomes of all the species that are represented within the input data. In reality, this end result is still very difficult to achieve.
Metagenomic assembly is further complicated by:
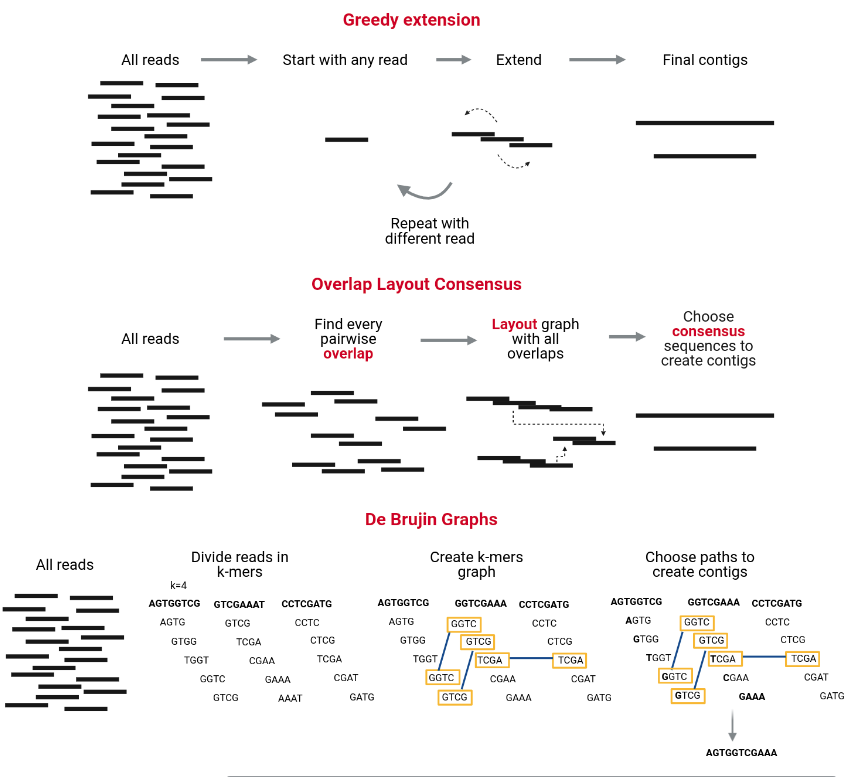
For assembly, there are 3 main strategies:
In this tutorial, we will learn how to run metagenomic assembly tool and evaluate the quality of the generated assemblies.
We will use data from the study: Temporal shotgun metagenomic dissection of the coffee fermentation ecosystem.
For an in-depth analysis of the structure and functions of the coffee microbiome, a temporal shotgun metagenomic study (six time points) was performed. The six samples have been sequenced with Illumina MiSeq utilizing whole genome sequencing.
As explained before, there are many challenges to metagenomics assembly, including:
To reduce the differences in coverage between samples, we can use a co-assembly approach, where reads from all samples are aligned together:
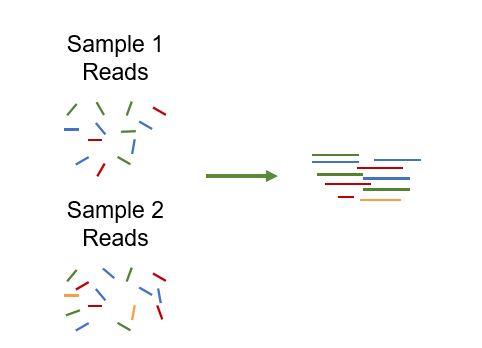
| Pros of co-assembly | Cons of co-assembly |
|---|---|
| More data | Higher computational overhead |
| Better/longer assemblies | Risk of shattering your assembly |
| Access to lower abundant organisms | Risk of increased contamination |
Co-assembly is then not always beneficial:
Co-assembly is reasonable if:
If it is not the case, individual assembly should be preferred. In this case, an extra step of de-replication should be used
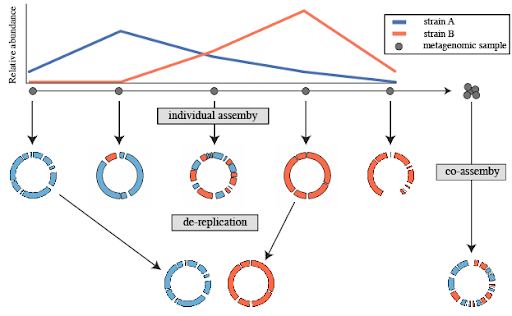
Co-assembly is more commonly used than individual assembly and then de-replication after binning. But in this tutorial, to show all steps, we will run an individual assembly.
NOTE! Sometimes it is important to run assembly tools both on individual samples and on all pooled samples, and use both outputs to get the better outputs for the certain dataset.
Several tools are available for metagenomic assembly. But 2 are the most used ones:
Both tools are available in Galaxy. But currently, only MEGAHIT can be used in individual mode for several samples.
NOTE!! See also Spades and Unicycler
A typical assembly workflow consists of:
FastQC and Trim Galore! or Cutadapt)MEGAHIT or MetaSPAdesMetaQUAST or QuastVALETBowtie2 combined with MultiQC to aggregate the results.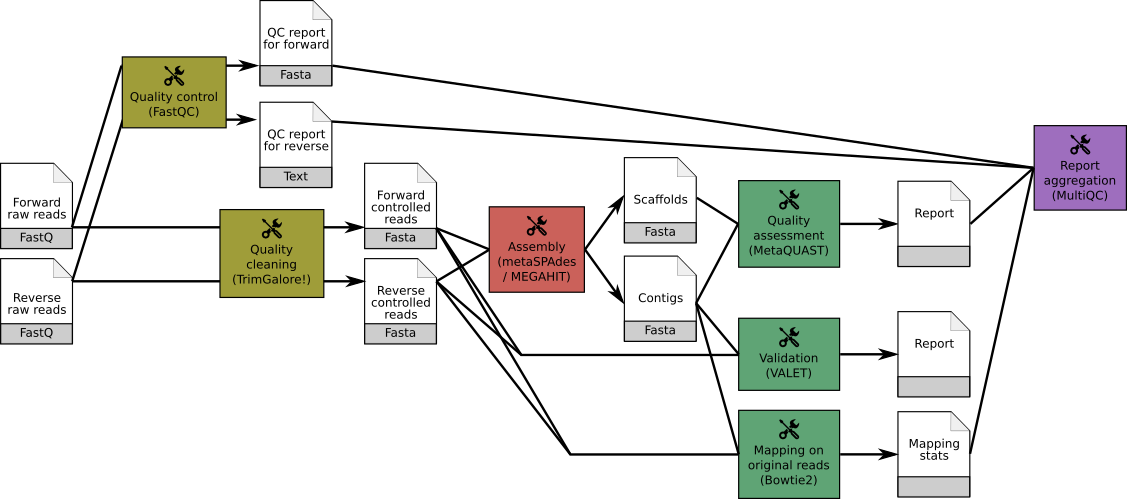
NOTE! Please, use:
https://zenodo.org/record/7818827/files/ERR2231567_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231567_2.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231568_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231568_2.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231569_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231569_2.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231570_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231570_2.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231571_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231571_2.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231572_1.fastqsanger.gz
https://zenodo.org/record/7818827/files/ERR2231572_2.fastqsanger.gz
MEGAHIT Tool with parameters
https://zenodo.org/record/7818827/files/contigs_ERR2231567.fasta
https://zenodo.org/record/7818827/files/contigs_ERR2231568.fasta
https://zenodo.org/record/7818827/files/contigs_ERR2231569.fasta
https://zenodo.org/record/7818827/files/contigs_ERR2231570.fasta
https://zenodo.org/record/7818827/files/contigs_ERR2231571.fasta
https://zenodo.org/record/7818827/files/contigs_ERR2231572.fasta
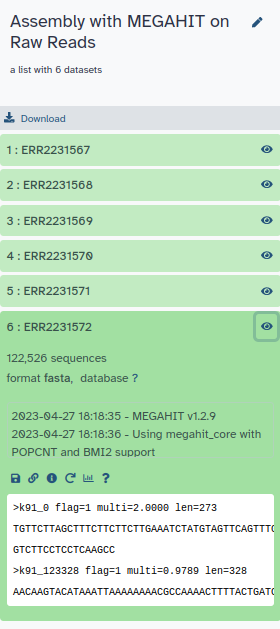
MEGAHIT produced a collection of output assemblies - one per sample -
that can be proceeded further in binning step and then de-replication.
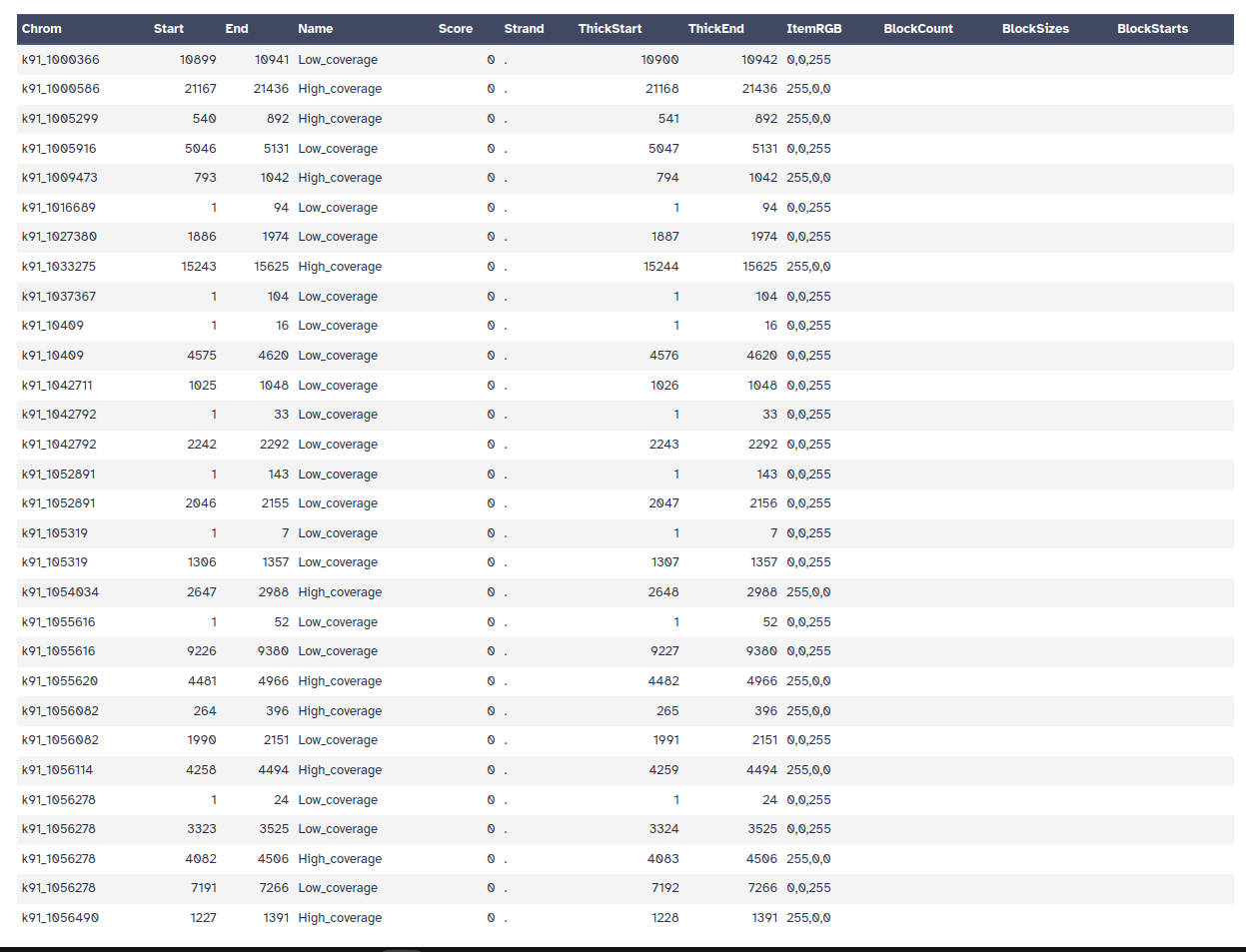
The output contains contigs, contiguous lengths of genomic sequences in which bases are known to a high degree of certainty.
Contrary to MetaSPAdes,
MEGAHIT does not output scaffolds, i.e. segments of genome sequence reconstructed from contigs and gaps.
The gaps occur when reads from the two sequenced ends of at least one fragment overlap with other reads from two different contigs (as long as the arrangement is otherwise consistent with the contigs being adjacent). It is possible to estimate the number of bases between contigs based on fragment lengths.
Question
MEGAHIT Tool with parameters
MetaSPAdes Tool with following parameters
NOTE! Slower than MEGAHIT
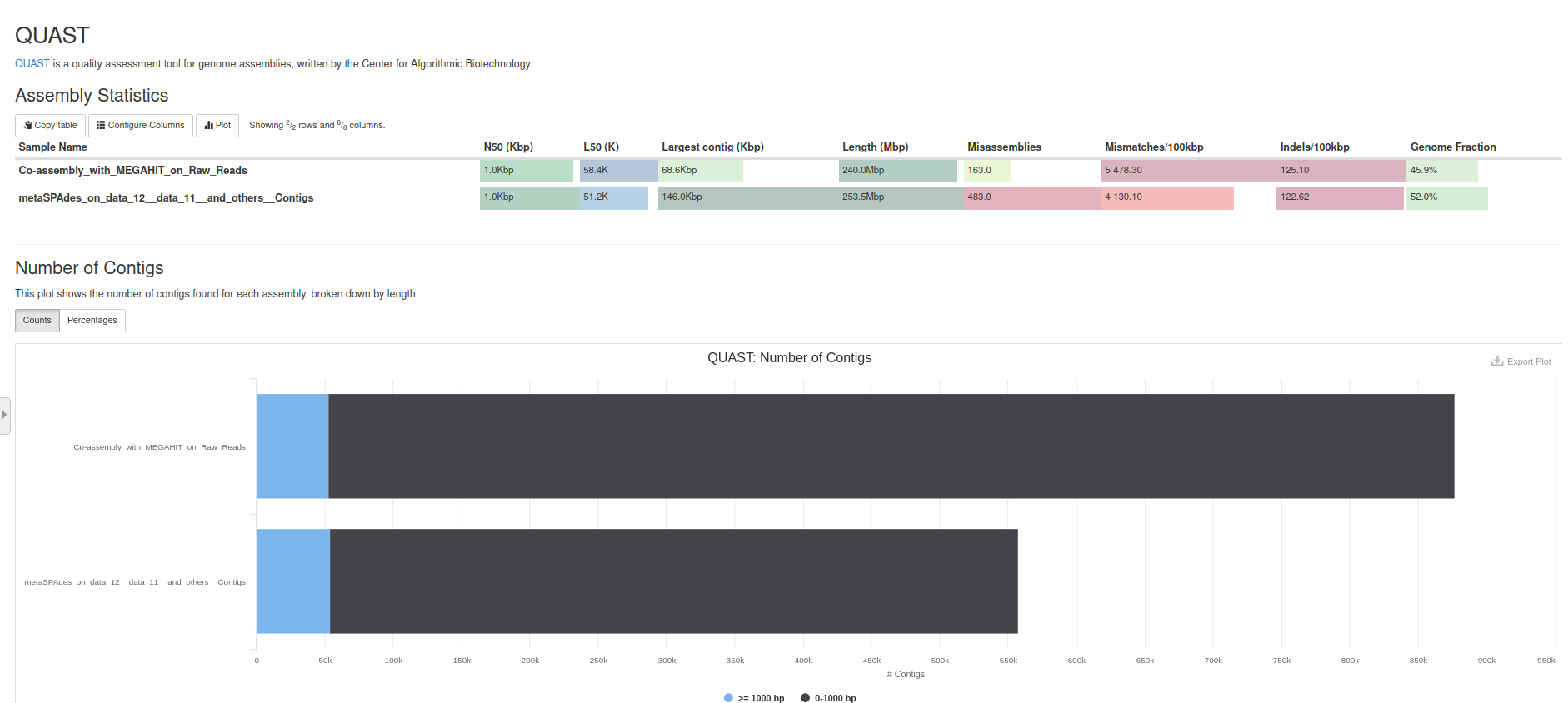
Quast Tool with parameters
MEGAHIT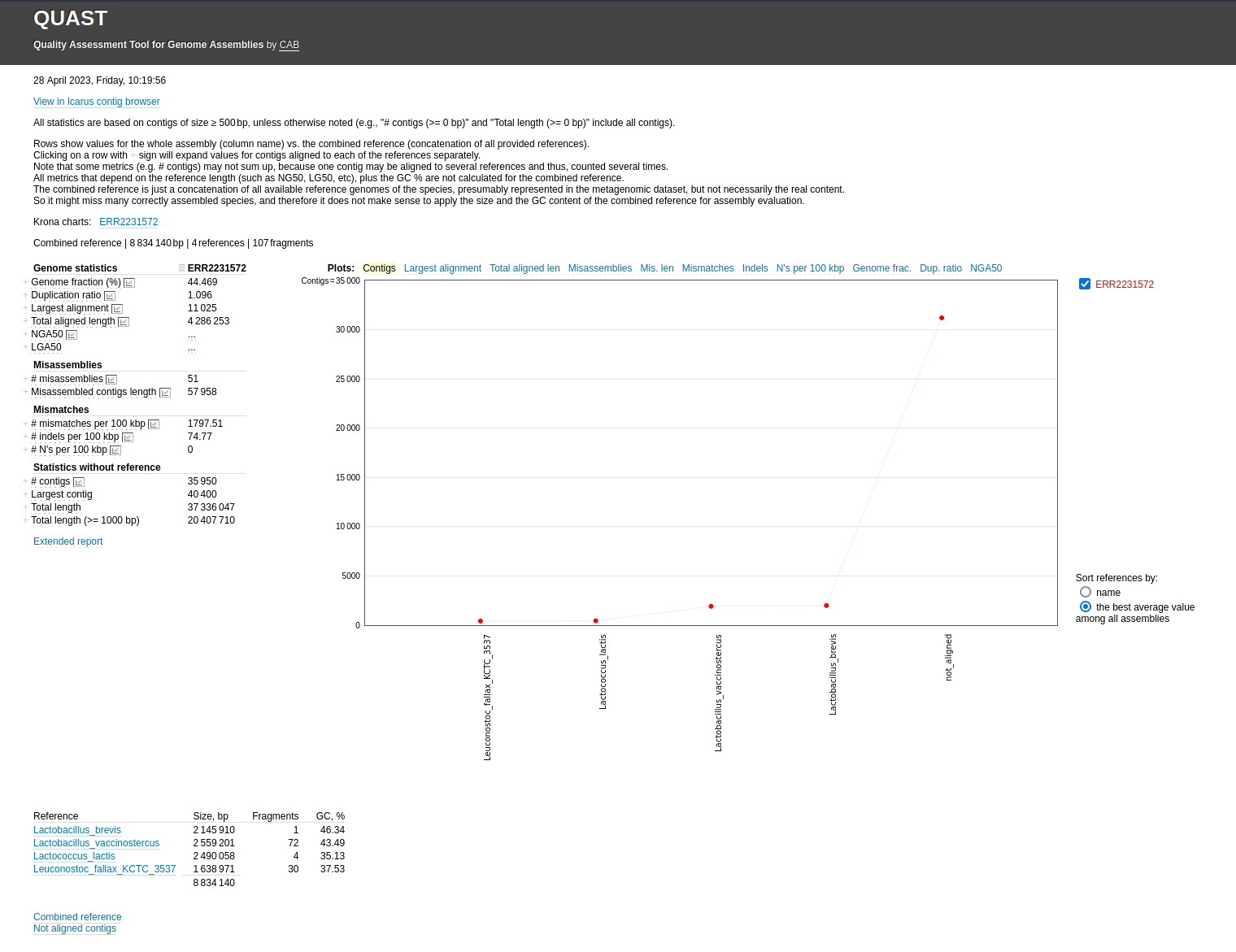
See Quast Manual
Genome fraction (%): percentage of aligned bases in the reference genome
Question
Duplication ratio: total number of aligned bases / genome fraction * reference length
Question
CoverM-CONTIG Tool with parameters:
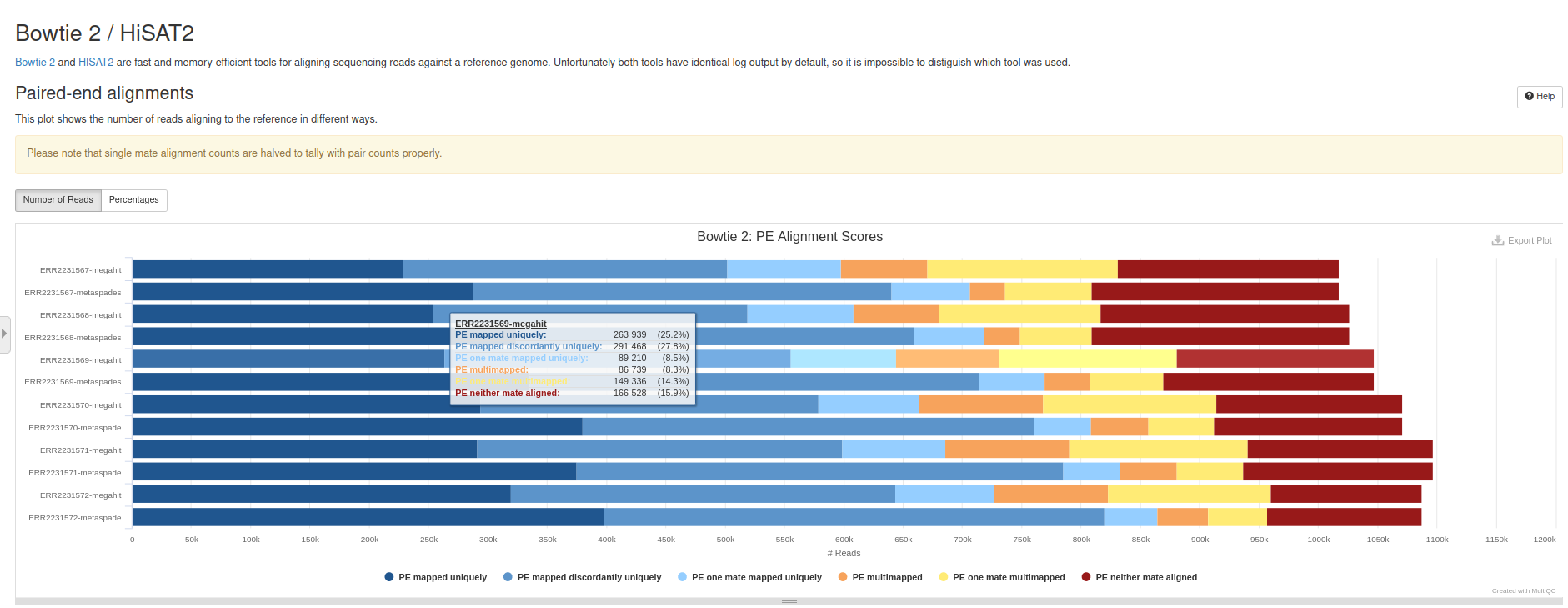
Bowtie2 Tool with the following parameters:
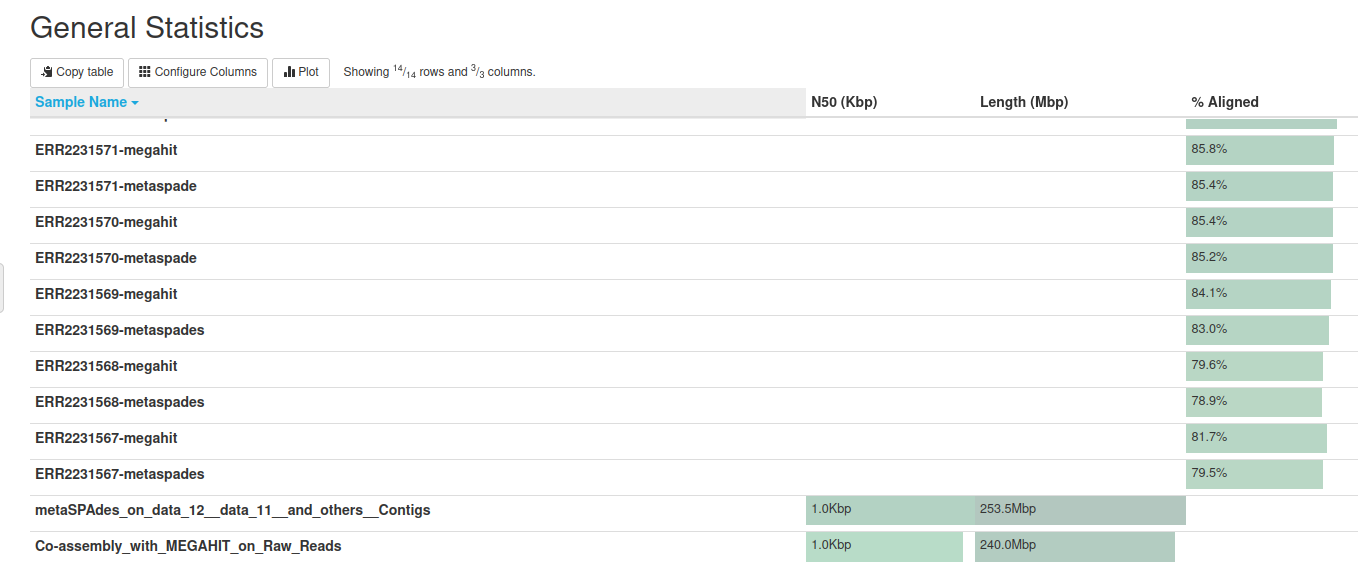
N50: length for which the collection of all contigs of that length or longer covers at least half an assembly
N50 statistic defines assembly quality in terms of contiguity. If all contigs in an assembly are ordered by length, the N50 is the minimum length of contigs that contains 50% of the assembled bases. For example, an N50 of 10,000 bp means that 50% of the assembled bases are contained in contigs of at least 10,000 bp.
When comparing N50 values from different assemblies, the assembly sizes must be the same size in order for N50 to be meaningful.
L50: number of contigs equal to or longer than N50
In other words, L50 is the minimal number of contigs that cover half the assembly.
Icarus generates contig size viewer and one or more contig alignment viewers (if reference genome/genomes are provided) that are accessible from the HTML report, by clicking on View on Icarus contig browser.
This viewer draws contigs ordered from longest to shortest. Let’s inspect this viewer for ERR2231568.
ERR22315680 and end as 500000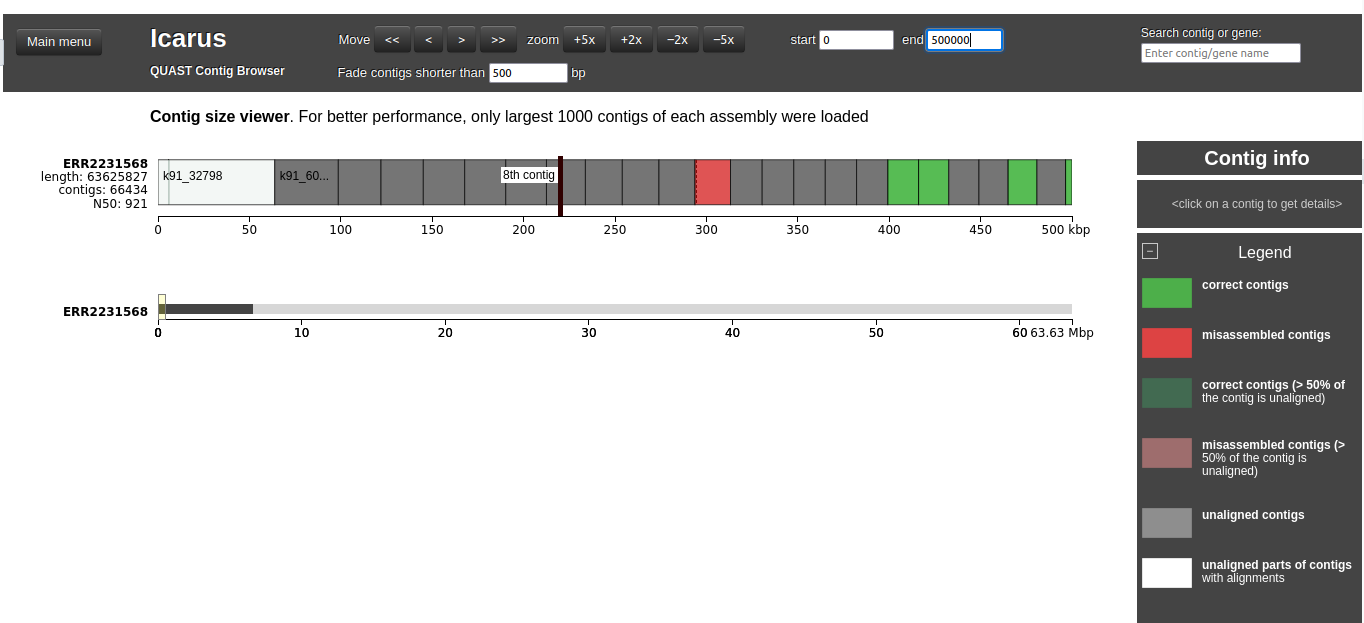
Question
Solutions
The first contig is white because >50% of the contigs is unaligned. By clicking on the contig, we see that only a small block is aligned:
223.41 – 223.65 kbp to Leuconostoc_pseudomesenteroides_KCTC_3652_NZ_BMBP01000002.1.
The red contig is a missamblied contig: it contains 2 blocks, with a translocation between them.
Click Main menu on the top left to go back to the main Icarus page.
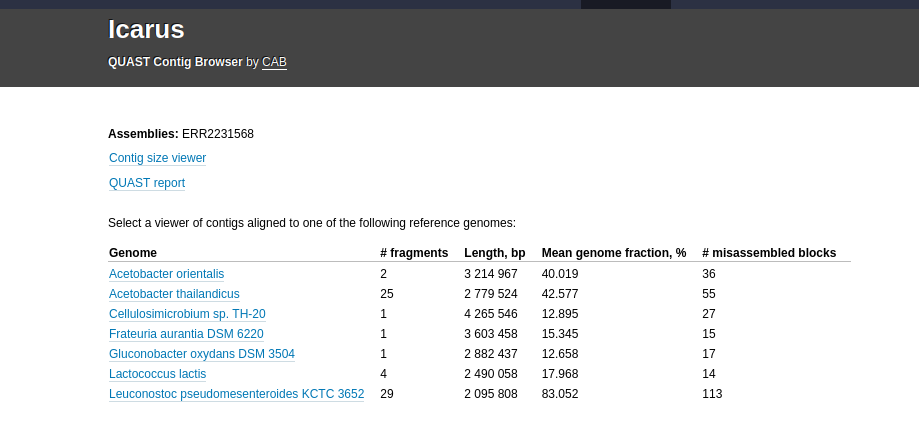
If a reference genome is provided, there should be a table on the main Icarus page that looks like:
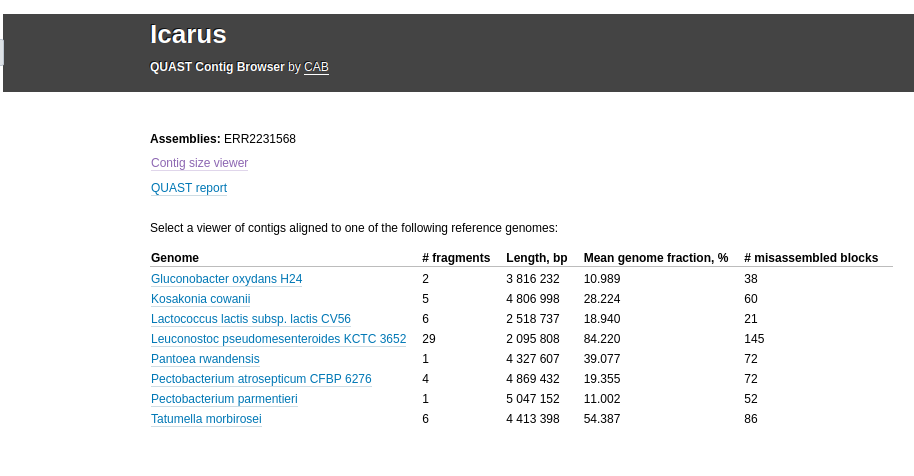
When clicking on the genome name, the contigs are displayed according to their mapping to the reference genome. The viewer can additionally visualize genes, operons, and read coverage distribution along the genome, if any of those were fed to QUAST.

Question
Solutions
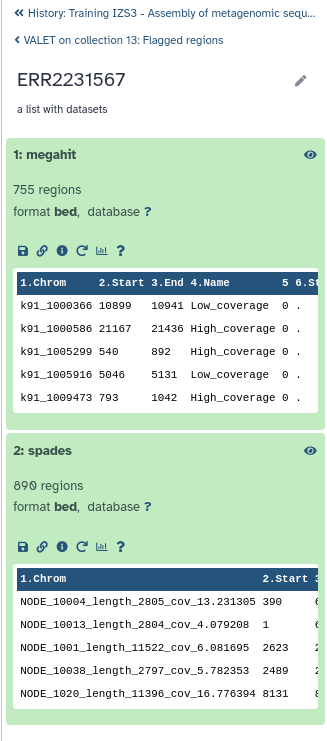
VALET (VALidating mETagenomic assemblies) is a pipeline for performing de novo validation of metagenomic assemblies.
VALET checks a number of properties that should hold true for a correct assembly
(e.g., mate-pairs are aligned at the correct distance from each other in the assembly,
the depth of coverage is fairly uniform along contigs, etc.).
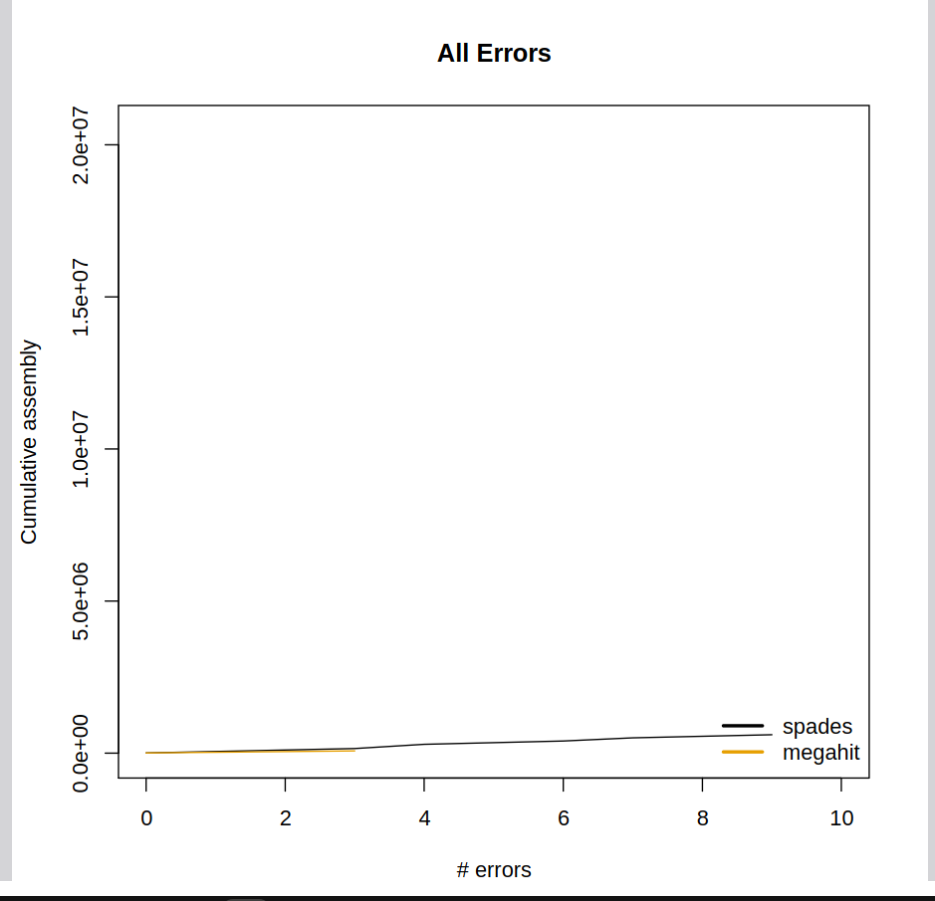
The violations of these invariants are reported allowing one to pinpoint areas that were potentially mis-assembled,
or to compare the quality of different assemblies.
For comparing multiple assemblies of the same data-sets,
VALET also reports an overall estimate of the likelihood a particular assembly is correct.
VALET Tool with paramaters:
metaSpadesMEGAHIT
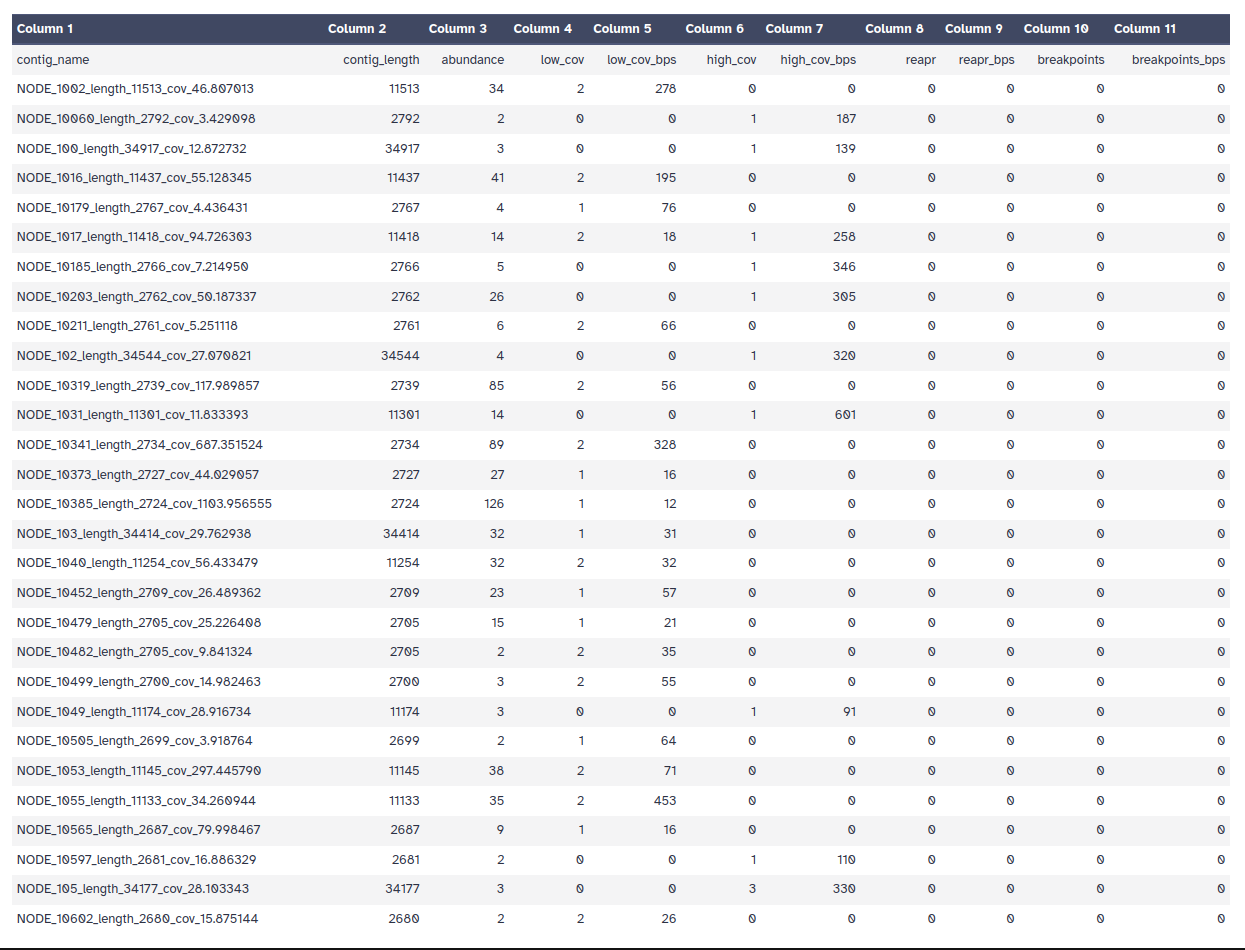
Bowtie2 on metaSpades outputBowtie2 on MEGAHIT outputQUAST on metaSpades outputQUAST on MEGAHIT outputMultiQC tool with following parameters

Metagenomic data can be assembled to, ideally, obtain the genomes of the species that are represented within the input data. But metagenomic assembly is complex and there are
MetaSPAdes and MEGAHITNow, it’s your turn!