How to identify pathogens using sequencing data? How to track the found pathogens through all your samples datasets?
Metagenomic analysis plays a pivotal role in unraveling the complex microbial communities present in various biological samples. One crucial aspect of metagenomics is the detection of pathogens within these communities, which holds significant implications for public health, clinical diagnostics, epidemiology, and environmental monitoring.
Detecting pathogens within metagenomic datasets is of paramount importance for several reasons:
Despite its significance, pathogen detection in metagenomics presents several challenges:
After detecting potential pathogens, it’s crucial to interpret the results accurately. This involves distinguishing between commensal and pathogenic strains, often achieved through comparative genomics and reference databases.
The applications of pathogen detection in metagenomic analysis are diverse and include:
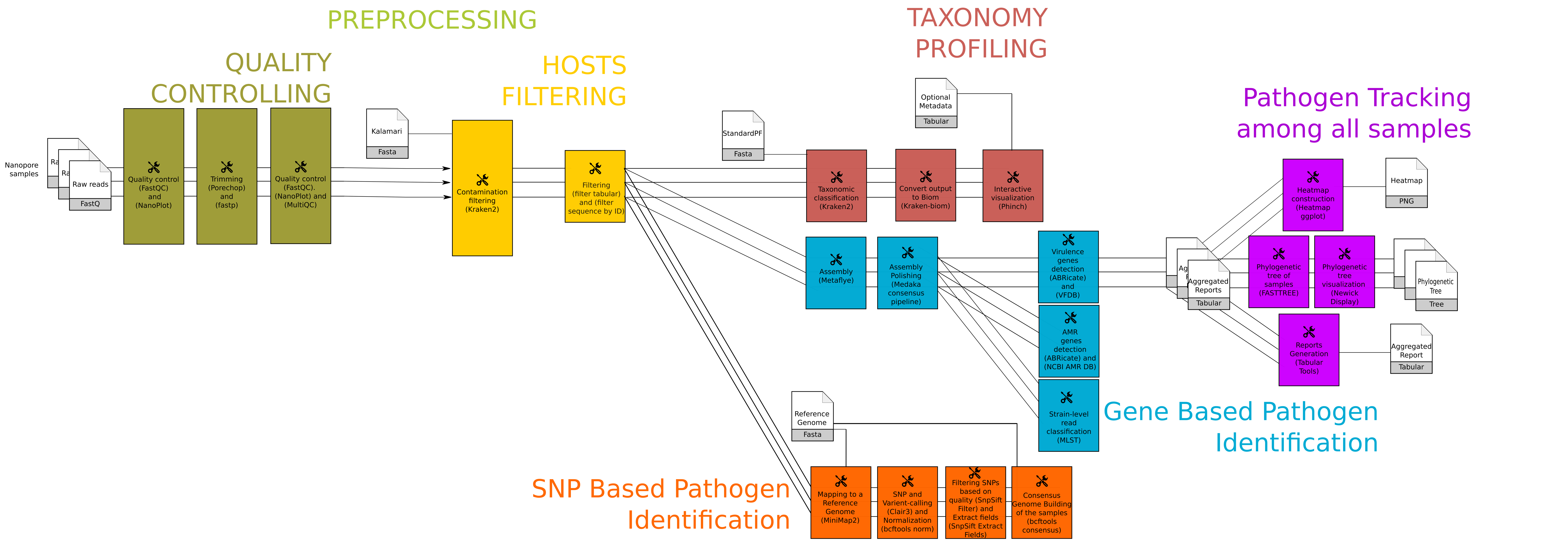
In this tutorial, we will be using datasets sourced from a comprehensive study that employed bioinformatics approaches to analyze whole-genome sequence data obtained from Vibrio cholerae isolates.
This study aimed to provide a complete array of virulence genes, pathogenicity islands, and antimicrobial resistance genes within these isolates.
The research holds particular significance as it sheds light on the genomic makeup of V. cholerae strains associated with cholera outbreaks in Uganda.
:NOTE! Please, use https://usegalaxy.eu/
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871245.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871246.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871247.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871248.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871249.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871250.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871251.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871252.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871253.fasta
https://github.com/vappiah/bioinfodata/raw/main/genome-assemblies/vibrio-cholerae/raw/batch1/SRR6871254.fasta
We will explore two distinct approaches for AMR gene detection, each based on specialized tools:
In order to search AMR genes among our samples’ contigs, we run ABRicate and choose the
NCBI Bacterial Antimicrobial Resistance Gene Database (AMRFinderPlus)
from the advanced options of the tool.
ABRicate checks if there is an AMR found or not,
if found then in which contig it is,
its location on the contig,
what the name of the exact product is,
what substance it provides resistance against and a lot of other information regarding the found AMR.
ABRicate Tool with the following parameters:
The outputs of ABRicate is a tabular file with different columns:
FILE: The filename this hit came fromSEQUENCE: The sequence in the filenameSTART: Start coordinate in the sequenceEND: End coordinateSTRAND: AMR geneGENE: AMR geneCOVERAGE: What proportion of the gene is in our sequenceCOVERAGE_MA: A visual represenationGAPS: Was there any gaps in the alignment - possible pseudogene?%COVERAGE: Proportion of gene covered%IDENTITY: Proportion of exact nucleotide matchesDATABASE: The database this sequence comes fromACCESSION: The genomic source of the sequencePRODUCT: Gene product (if available)RESISTANCE: Putative antibiotic resistance phenotype, ;-separated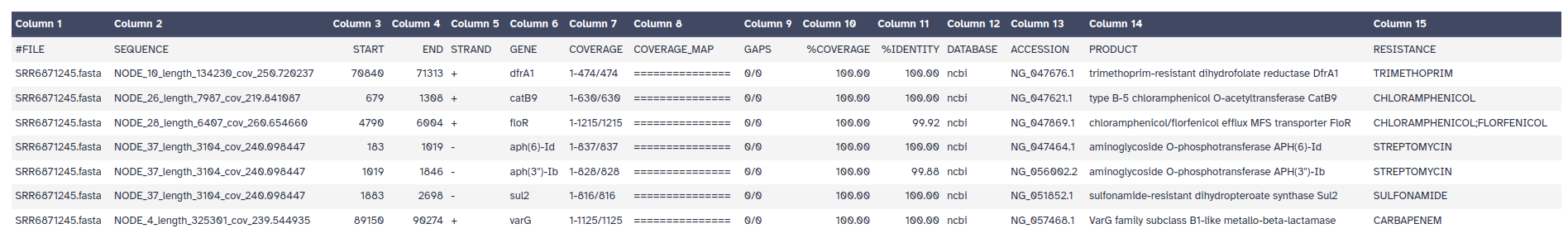
TRY! Search ACCESSION value on NCBI webserver
ABRicate can combine results into a simple matrix of gene presence/absence.
.%COVERAGE.
This can be individual abricate reports, or a combined one.ABRicate Summary Tool with the following parameters:
ABRicate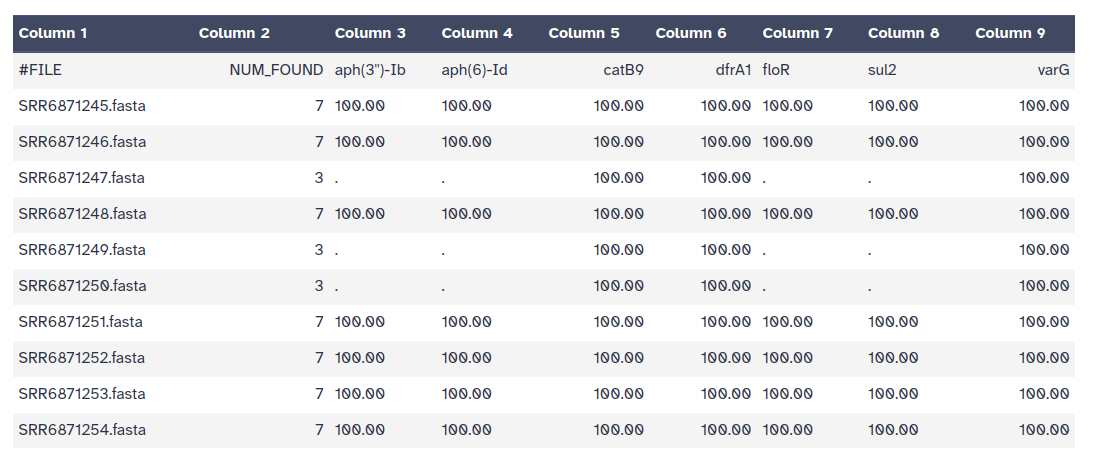
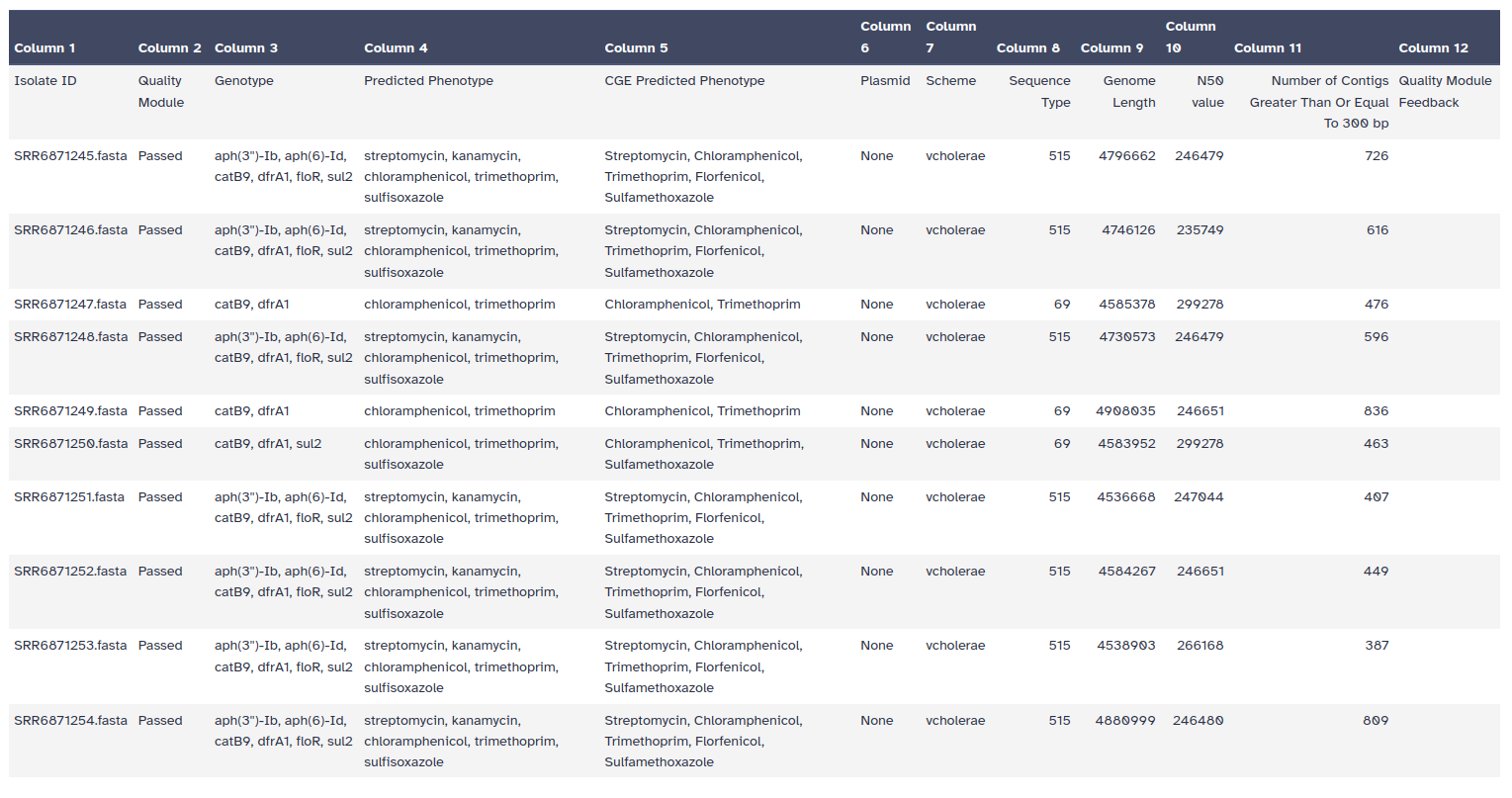
StarAMR scans bacterial genome contigs against both the ResFinder, PointFinder (Zankari et al. 2017),
and PlasmidFinder (used by the ResFinder webservice) and compiles a
summary report of detected antimicrobial resistance genes.
StarAMR Tool with the following parameters:
There are 5 different output files produced by StarAMR
The summary file is most important and provides all the resistance genes found.
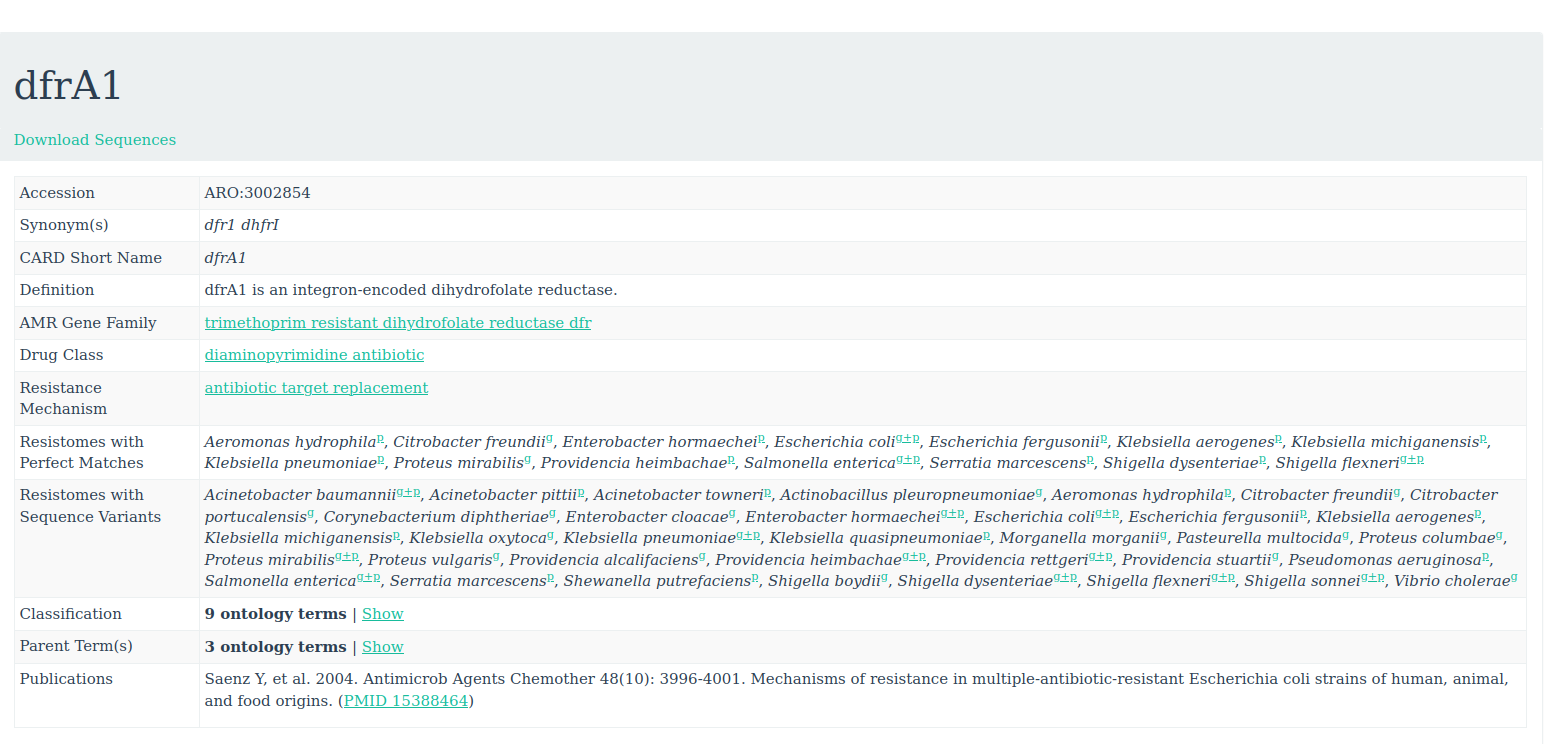
To get more information about these antibiotic resistant genes, you can check the CARD database (Comprehensive Antibiotic Resistance Database)
In this step we return back to the main goal of the tutorial where we want to identify the pathogens: identify if the bacteria found in our samples are pathogenic bacteria or not.
DEFINITIONS
To identify VFs, we use again ABRicate but this time with the VFDB from the advanced options of the tool.
ABRicate Tool with the following parameters:
Rename output collection as VFs
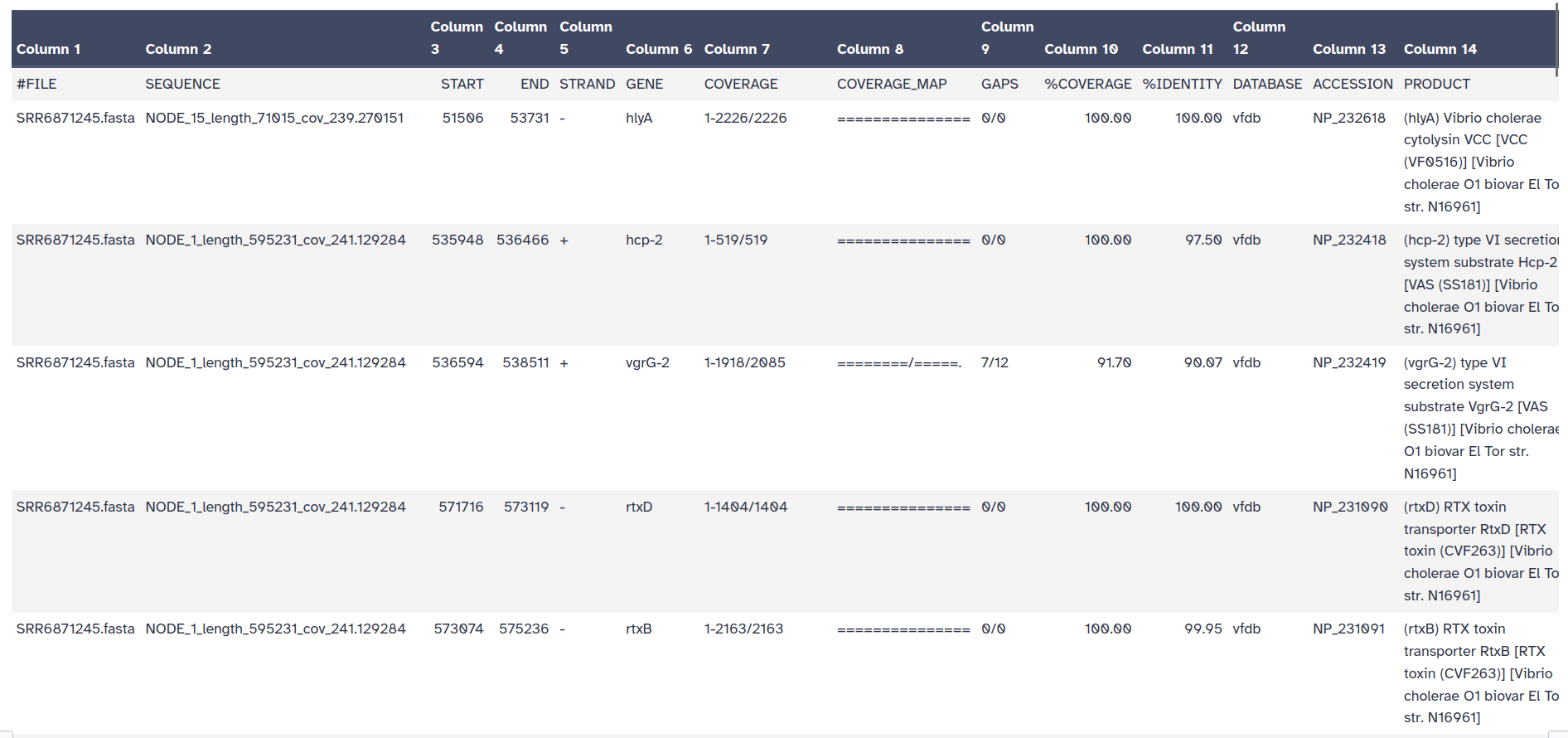
ABRicate Summary Tool with the following parameters:
ABRicate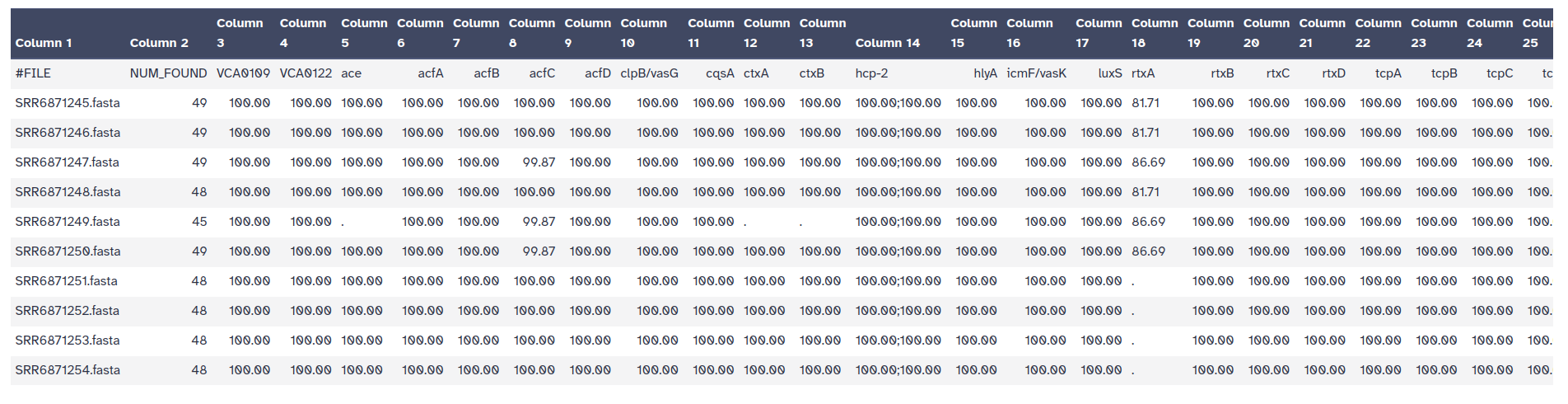
In this last section, we would like to show how to aggregate results and use the results to help tracking pathogenes among samples by:
With these two types of visualizations we can have an overview of all samples and the genes, but also how samples are related to each other i.e. which common pathogenic genes they share.
Given the time of the sampling and the location one can easily identify using these graphs, where and when the contamination has occurred among the different samples.
To execute the analysis in this section,
it is essential to handle and format the ABRicate output.
This processing is aimed at extracting a curated list comprising
Accession IDs of identified genes associated with Virulence Factors, along with their corresponding sample IDs.
Cut Tool with the following parameters:
Cut Tool with the following parameters:
Unique occurrences of each record Tool with the following parameters:
CutConcatenate multiple datasets (tail-to-head by specifying how Tool with the following parameters:
A heatmap is one of the visualization techniques that can give you a complete overview of all the samples together and whether or not a certain value exists. In this tutorial, we use the heatmap to visualize all samples aside and check which common bacteria pathogen genes are found in samples and which is only found in one of them.
We use Heatmap w ggplot tool along with other tabular manipulating tools to prepare the tabular files.
1 Combine VFs accessions for samples into a table and get 0 or 1 for absence / presence
Collapse Collection Tool with the following parameters:
Add line to file Tool with the following parameters:
Collapse CollectionMulti-Join Tool with the following parameters:
Add line to fileReplace Tool with the following parameters:
Multi-Joindataset_(.*?)_Sample_(\S+)Acc_$1Replace Tool with the following parameters:
Replace toolAcc_00Replace Tool with the following parameters:
ReplaceAcc_\S*1Advanced Cut Tool with the following parameters:
Multi-Join toolAdvanced Cut Tool with the following parameters:
Replace toolPaste Tool with the following parameters:
Advanced Cut (Tool N. 7)Advanced Cut (Tool N. 8)Delimit by:Tab2 Draw heatmap
Transpose Tool with the following parameters:
PasteHeatmap w ggplot Tool with the following parameters:
Transpose tool
Phylogenetic trees can be used to track the evolution of the pathogen between the samples. Therefore, the VFs are used as a marker gene for the pathogen, similar to 16S marker genes for species profiling.
We use the VFs since we know they are associated to the pathogenicity of the sample.
By observing the created trees one can identify groups of related pathogens. If additional meta data of the samples would be available one could further identify groups that are associated to specific traits such as increase pathogenicity or faster transmission.
Consequently, the tree could be used for phylogenetic placement of unknwon samples.
For the phylogenetic trees,
for each bacteria pathogen gene found in the samples we use ClustalW for Multiple Sequence Alignment (MSA)
needed before constructing a phylogenetic tree,
for the tree itself we use FASTTREE and Newick Display to visualize it.
1 Extract the sequences of the VFs
To get the sequence to align, we need to extract the sequences of the VFs in the contigs:
Collapse Collection Tool with the following parameters:
Collapse Collection Tool with the following parameters:
Split by group Tool with the following parameters:
Collapse Collection toolRemove beginning Tool with the following parameters:
Split by groupFilter sequences by ID Tool with the following parameters:
Collapse Collection toolSplit by group tool2 Phylogenetic Tree building
We can now run multiple sequence alignment, build the trees for each VF and display them.
ClustalW Tool with the following parameters:
Filter sequences by ID toolFASTTREE Tool with the following parameters:
ClustalW toolNewick Display Tool with the following parameters:
FASTTREENB It takes a lot of time!!!
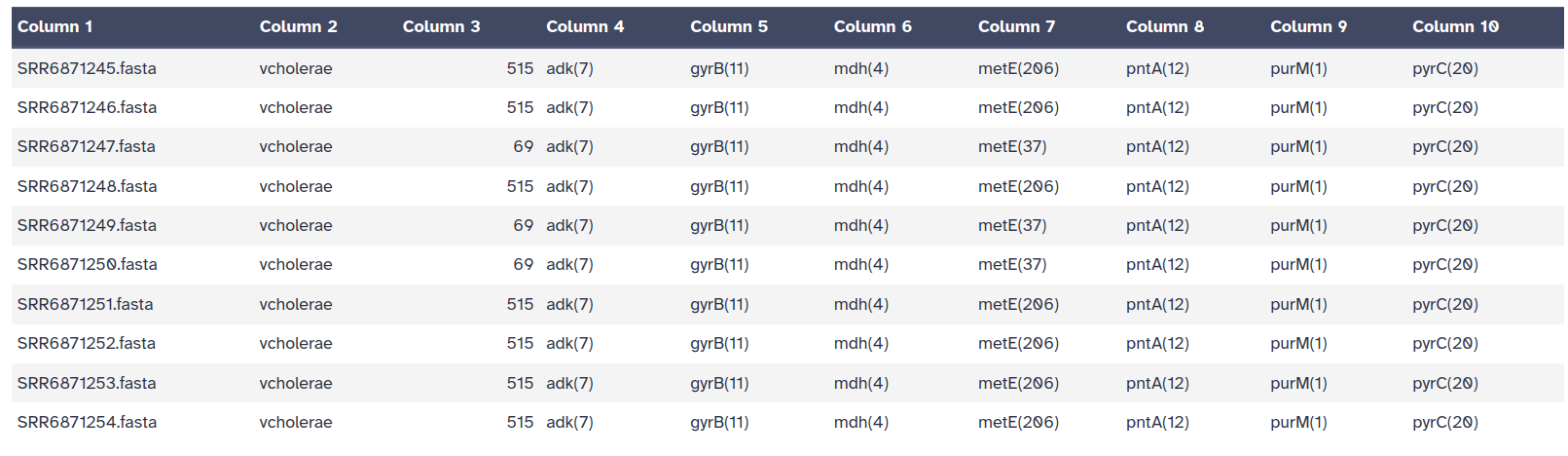
MLST tool is used to scan the pubMLST database against PubMLST typing schemes.
It’s one of the analyses that you can perform on your dataset to determine the allele IDs,
you can also detect novel alleles.
This step is not essential to identify pathogens and track them in the remainder of this tutorial, however we wanted to show some of the analysis that one can use Galaxy in to understand more about the dataset, as well as identifying the strain that might be a pathogen or not.
MLST Tool with the following parameters:
The output file of the MLST tool is a tab-separated output file which contains:
In this tutorial, we worked with isolated genomes.
To identify and track foodborne pathogens using metagenomic sequencing, different samples of potentially contaminated food (at different time points or different locations) are prepared, DNA is extracted and sequenced. The generated sequencing data then need to be processed using bioinformatics tools.
Generally, we are not interested in the food (host) sequences, rather only those originating from the pathogen itself.
It is an important to get rid of all host sequences and to only retain sequences that might include a pathogen, both in order to speed up further steps and to avoid host sequences compromising the analysis.
In this part of tutorial we will use samples from chicken meat spiked with Salmonella
Kraken2 and Kalamari, a database of completed assemblies for metagenomics-related tasks used widely in contamination and host filteringKraken2 assignments
Kraken2 classification to extract the sequence ids of all hosts sequences identified with Kraken2NOTE! Please, use
https://zenodo.org/record/7593928/files/Barcode10_Spike2.fastq.gz
https://zenodo.org/record/7593928/files/Barcode11_Spike2b.fastq.gz
Kraken2 Tool with the following parameters:
Kraken2 collectionQuestion
Krakentools: Convert kraken report file Tool with the following parameters:
Kraken2Krona pie chart Tool with the following parameters:
Krakentools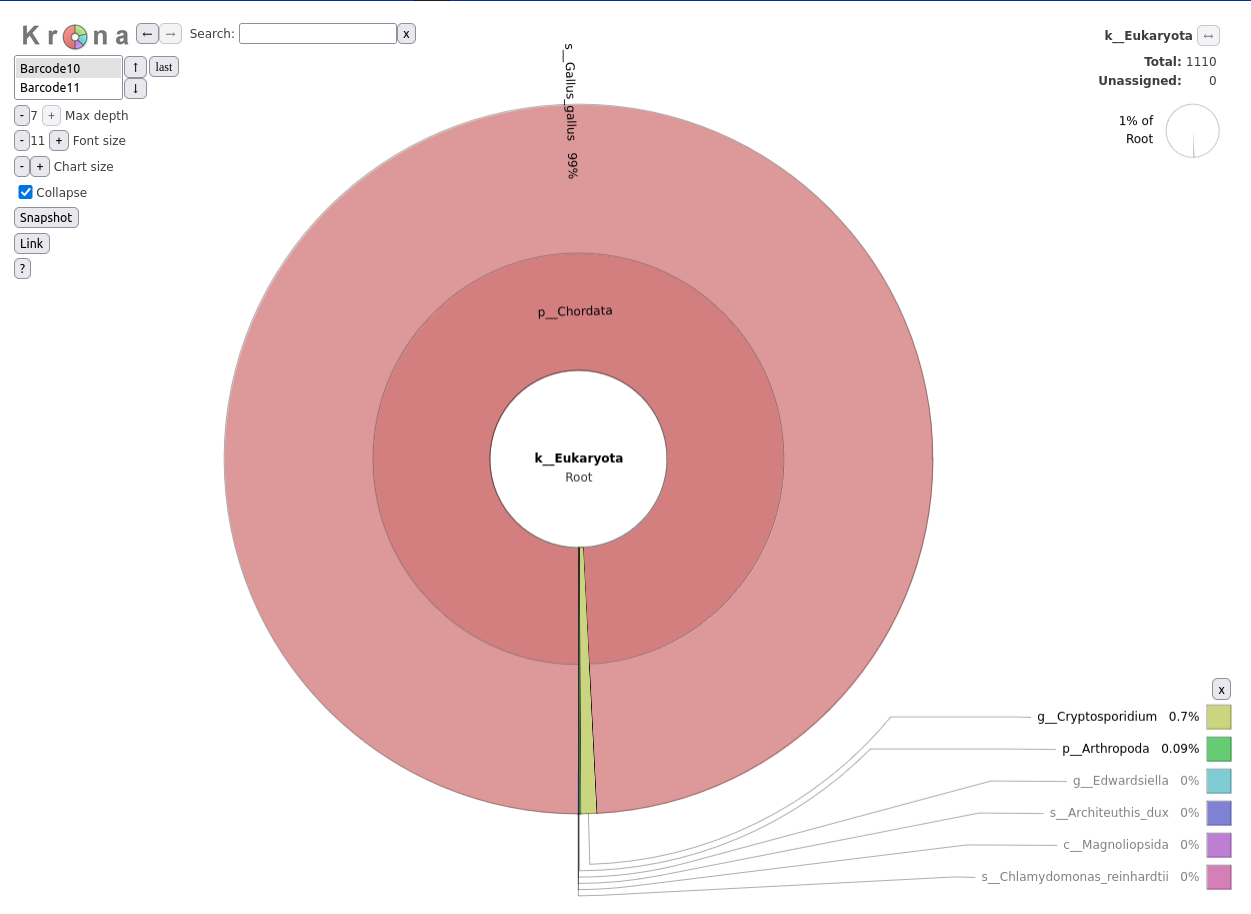
Krakentools: Extract Kraken Reads By ID Tool with the following parameters:
Kraken2 toolKraken2 toolTaxonomix ID(s) to match:9031 9606 9913
We specify here the taxonomic ID of the hosts so we can filter reads assigned to these hosts. Kraken2 uses taxonomic IDs from NCBI, the IDs for a specific taxa can be found at ncbi. To be generic, we remove here (if the contaminated food comes from and may include other animals, you can change the values):
Now, it’s your turn!