It explains how to detect somatic mutations in cancer samples, which variant callers are available, and how to customize the detection process.
Starting from BAM files, Musta efficiently performs variant calling in either tumor-normal matched mode or tumor-only mode, depending on the provided input files for each sample. To ensure the highest accuracy, Musta utilizes a combination of six powerful variant callers:
To achieve a refined and accurate consensus, an ensemble approach is employed, utilizing the machine learning-based algorithm of SomaticSeq.
The Detection module within Musta provides users with the capability to initiate variant calling processes. To execute this module, you can use the following basic Musta command structure:
musta detect [OPTIONS]
Here’s breakdown of each option available in the Musta Detection module:
General Options:
-h, --help: Show the help message and exit.Required Options:
--workdir PATH (-w PATH): Specifies the destination folder for analysis. This is where the Snakemake pipeline, logs, and analysis outputs will be located.--samples-file PATH (-s PATH): Points to a YAML file listing the datasets you wish to analyze. This file provides information about the samples to be processed. For further details, please refer to the dedicate section.--reference-file PATH (-r PATH): Identifies the path to the reference FASTA file, which is essential for the analysis. For further details, please refer to the dedicate section.--bed-file PATH (-b PATH): Points to a compressed and indexed BED file. This file lists the regions that the analysis should be restricted to. For further details, please refer to the dedicate section.Optional Options:
--variant-file PATH (-v PATH): Specifies the path to a VCF file containing variants and allele frequencies (used only when running Mutect). For further details, please refer to the dedicate section.--germline-resource PATH (-g PATH): Indicates the path to a population VCF of germline sequencing data. This file contains allele fractions and is used only when running Mutect. For further details, please refer to the dedicate section.--dbsnp-file PATH (-db PATH): Specifies the path to a compressed and indexed VCF file containing known germline variants. This option is used exclusively when running LoFreq and/or MuSe.For further details, please refer to the dedicate section.--tmpdir PATH (-t PATH): Specifies the path to the temporary directory. Ensure that this directory has sufficient storage capacity to hold the intermediate files generated during the analysis.Variant Caller Exclusion Options:
--exclude-mutect (-emu): Excludes the Mutect variant caller from the analys.--exclude-lofreq (-elf): Excludes the LoFreq variant caller from the analysis.--exclude-strelka (-esk): Excludes the Strelka variant caller from the analysis.--exclude-muse (-ems): Prevents the MuSe variant caller from running.--exclude-varscan (-evs): Excludes the VarScan variant caller from the analysis.--exclude-vardict (-evd): Prevents the VarDict variant caller from running.Variant Caller Filtering Options:
--strict: Runs only the restrictive variant callers, which include Mutect, LoFreq, and Strelka.--soft: Runs only the permissive variant callers, which include VarScan, VarDict, and MuSe.--fast: Executes only the fast variant callers, which are LoFreq, VarScan, and Strelka.Additional Options:
--force, -f: Forces the re-creation of all output files.--dryrun, -d: Describes the workflow but does not execute it. This is useful for previewing the analysis steps before running them.--summary-only, -so: Generate summary reports without re-running the analysis.Obtain the “test-data-somatic” Dataset: First, download the “test-data-somatic” dataset by following the instructions provided in the dedicated section of this user guide.
Create an Input Samples YAML File:
Prepare an input samples.yml file, specifying your sample names, normal and tumor sample information, and paths to the corresponding BAM files. Here’s an example YAML structure:
sample_A25:
normal_sample_name:
- N1
tumor_sample_name:
- A25
normal_bam:
- /path/to/test-data-somatic/data/bam/N1.chr22.bam
tumor_bam:
- /path/to/test-data-somatic/data/bam/A25.chr22.bam
sample_B33:
normal_sample_name:
- N1
tumor_sample_name:
- B33
normal_bam:
- /path/to/test-data-somatic/data/bam/N1.chr22.bam
tumor_bam:
- /path/to/test-data-somatic/data/bam/B33.chr22.bam
sample_C2:
normal_sample_name:
- N1
tumor_sample_name:
- C2
normal_bam:
- /path/to/test-data-somatic/data/bam/N1.chr22.bam
tumor_bam:
- /path/to/test-data-somatic/data/bam/C2.chr22.bam
Modify it according to your specific requirements.
Run Musta Detect with All Variant Callers (Excluding Strelka):
Execute the following command to run the “Musta Detect” module with all variant callers enabled, excluding Strelka, using the “test-data-somatic” dataset. Strelka has been excluded from this run due to compatibility issues with the reference and support files, which have been reduced to include only chromosome 22. If you wish to include Strelka in your analysis, you should use the original reference and support files for the full genome
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/test-data-somatic/resources/chr22.fa \
--bed-file /path/to/test-data-somatic/resources/chr22.sorted.bed.gz \
--germline-resource /path/to/test-data-somatic/resources/somatic-b37_af-only-gnomad.raw.sites.ucsc.chr22.vcf \
--variant-file /path/to/test-data-somatic/resources/somatic-b37_small_exac_common_3.ucsc.chr22.vcf \
--dbsnp-file /path/to/test-data-somatic/resources/dbsnp.chr22.vcf.gz \
--exclude-strelka
Ensure you replace the /path/to/ placeholders with the actual file paths on your system.
These steps will help you quickly initiate somatic mutation detection using the “test-data-somatic” dataset within the Musta Detect module.
After initiating the execution of musta detect, you will see the Musta log unfolding on your screen:
YYYY-MM-DD hh:mm:ss|INFO |main |musta v1.0.0 - End-to-end pipeline to detect, classify and interpret mutations in cancer
YYYY-MM-DD hh:mm:ss|INFO |main |Somatic Mutations Detection.
1. Multiple Variant Calling: mutect, lofreq, varscan, vardict, muse, strelka.
2. Ensemble consensus approach to combine results and to improve the performance of variant calling
YYYY-MM-DD hh:mm:ss|INFO |main |Reading configuration file
YYYY-MM-DD hh:mm:ss|INFO |main |Setting paths
YYYY-MM-DD hh:mm:ss|INFO |main |Deploying musta:v1.2.0.1 pipeline from https://github.com/solida-core/musta.git
YYYY-MM-DD hh:mm:ss|INFO |main |Initializing Config file
YYYY-MM-DD hh:mm:ss|INFO |main |Initializing Samples file
YYYY-MM-DD hh:mm:ss|INFO |main |Running
YYYY-MM-DD hh:mm:ss|INFO |main |Variant Calling
YYYY-MM-DD hh:mm:ss|INFO |main |Variant Caller: 'mutect'
Building DAG of jobs...
After a few minutes, you will see the last lines of the log, and the execution of Musta will conclude. You can locate the resulting analysis in the following destination folders:
These folders will contain the relevant data and reports generated during the Musta Detect execution.
Let’s take a look at the newly created <WORKDIR> folder
.
├── benchmarks
│ └── detection
├── logs
│ ├── detection
│ └── samplename
├── musta
│ ├── config
│ ├── LICENSE
│ ├── README.md
│ ├── resources
│ └── workflow
└── outputs
├── YYYYMMDD-hhmmss.report.html
├── bedfile
├── detection
└── samplename
─ benchmarks
│ └── detection
│ ├── lofreq
│ │ ├── sample_A25.lofreq.txt
│ │ ├── sample_B33.lofreq.txt
│ │ └── sample_C2.lofreq.txt
│ ├── muse
│ │ ├── ...
│ ├── mutect
│ │ ├── ...
│ ├── somatiqseq
│ │ ├── sample_A25.somaticseq.txt
│ │ ├── sample_B33.somaticseq.txt
│ │ └── sample_C2.somaticseq.txt
│ ├── vardict
│ │ ├── ...
│ └── varscan
│ ├── ...
detection: In this subdirectory, you’ll find log files related to the detection module. These logs provide detailed information about the execution of various variant callers.samplename: This subdirectory contains logs generated by GATK tools
── logs
│ ├── detection
│ │ ├── lofreq
│ │ │ ├── sample_A25.lofreq.log
│ │ │ ├── sample_B33.lofreq.log
│ │ │ └── sample_C2.lofreq.log
│ │ ├── muse
│ │ │ ├── ...
│ │ ├── mutect
│ │ │ ├── ...
│ │ ├── somaticseq
│ │ │ ├── sample_A25.somaticseq.log
│ │ │ ├── sample_B33.somaticseq.log
│ │ │ └── sample_C2.somaticseq.log
│ │ └── varscan
│ │ ├── ...
│ └── samplename
│ ├── sample_A25.gsn.normal.log
│ ├── sample_A25.gsn.tumor.log
│ ├── ...
musta: The directory houses the core of the Musta tool, including the Snakemake pipeline, which can be accessed from the official Musta GitHub repository: https://github.com/solida-core/musta.
YYMMDD-hhmmss.report.html: Auto-generated, independent, detailed HTML reports that include runtime statistics, provenance information, workflow topology, and results. As an example, the report of the Snakemake Musta Detection can be found here.bedfile: Within this subdirectory, you will find refactored BED files.samplename: This subdirectory contains outputs from GATK Tools, which are part of the analysis process.detection: The directory houses the outcomes and data derived from the execution of Musta’s detection module. Within this directory, users can explore a wealth of output files and data that are byproducts of the analysis. Specifically, you can find:
.
├── lofreq
├── muse
├── mutect
├── results
│ ├── sample_A25.musta.indels.vcf.gz
│ ├── sample_A25.musta.indels.vcf.gz.tbi
│ ├── sample_A25.musta.snvs.vcf.gz
│ ├── sample_A25.musta.snvs.vcf.gz.tbi
│ ├── sample_A25.somatic.lofreq.indels.vcf.gz
│ ├── sample_A25.somatic.lofreq.indels.vcf.gz.tbi
│ ├── sample_A25.somatic.lofreq.snvs.vcf.gz
│ ├── sample_A25.somatic.lofreq.snvs.vcf.gz.tbi
│ ├── sample_A25.somatic.muse.vcf.gz
│ ├── sample_A25.somatic.muse.vcf.gz.tbi
│ ├── sample_A25.somatic.mutect.vcf.gz
│ ├── sample_A25.somatic.mutect.vcf.gz.tbi
│ ├── sample_A25.somatic.vardict.vcf.gz
│ ├── sample_A25.somatic.vardict.vcf.gz.tbi
│ ├── sample_A25.somatic.varscan.indels.vcf.gz
│ ├── sample_A25.somatic.varscan.indels.vcf.gz.tbi
│ ├── sample_A25.somatic.varscan.snvs.vcf.gz
│ ├── sample_A25.somatic.varscan.snvs.vcf.gz.tbi
│ ├── sample_B33....
│ └── sample_C2....
├── somaticseq
├── vardict
└── varscan
The VCF files labeled <SAMPLE_NAME>.musta.*.vcf.gz represent the final results,
and they are produced by the SomaticSeq ensemble.
Within the results directory,
a dedicated summary folder has been organized to compile and present essential aggregated data.
This structured collection aids users in comprehending and interpreting key metrics and visual representations.
Below is a detailed list of the files available in the summary folder:
└── summary
├── common_pass_variants_heatmap.png
├── common_variants_counts.tsv
├── common_variants_mean.tsv
├── mean_pass_variants_plot.png
├── mean_pass_variants.tsv
├── mean_runtime_plot.png
├── mean_runtime.png
├── pass_variants_data.json
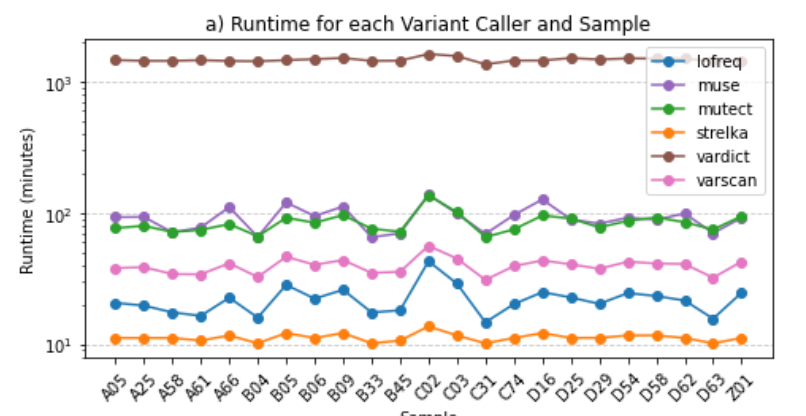
├── runtime_for_sample_and_variant_caller.png
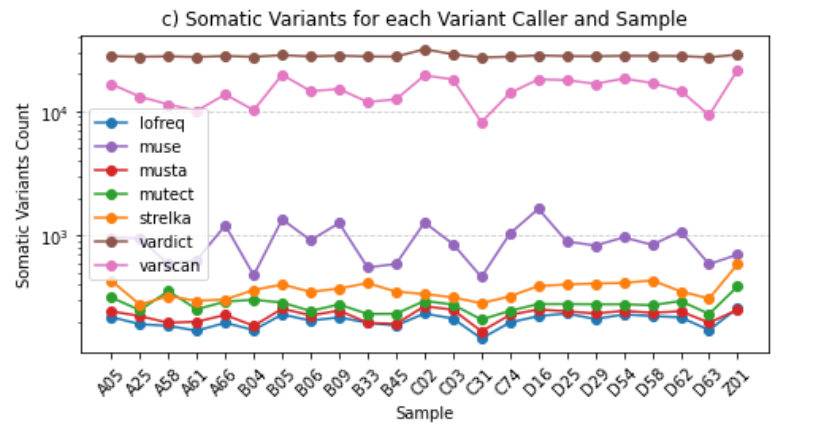
├── somatic_variants_for_sample_and_variant_caller.png
└── summary_for_each_sample_and_variant_caller.tsv
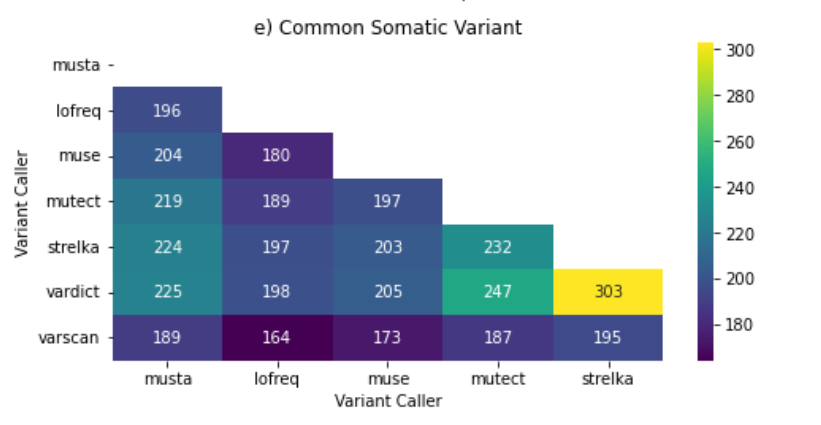
common_pass_variants_heatmap.png: Presents a heatmap illustrating the common pass variants across different variant callers.
common_variants_counts.tsv: Tabular data outlining the counts of common variants for each variant caller.
common_variants_mean.tsv: Tabular data providing the mean values of common variants for each variant caller.
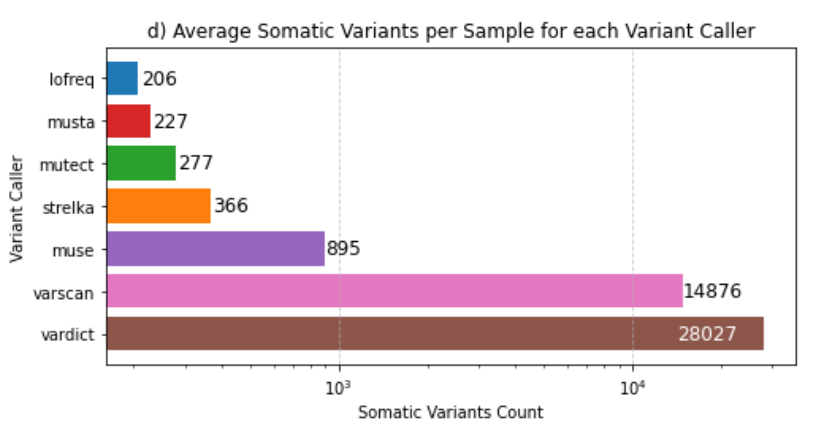
mean_pass_variants_plot.png: Visual representation of the mean pass variants, aiding in trend analysis.
mean_pass_variants.tsv: Tabular data offering insights into the mean pass variants for each variant caller.
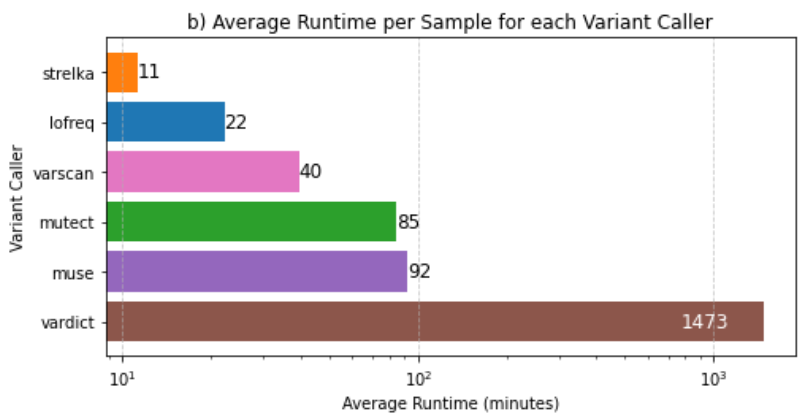
mean_runtime_plot.png: Graphical representation depicting the mean runtime across different variant callers.
mean_runtime.png: Tabular data presenting the mean runtime values for each variant caller.
pass_variants_data.json: JSON file storing pass variants for each sample and variant caller.
runtime_for_sample_and_variant_caller.png: Graphical representation of the runtime distribution for each sample and variant caller.
somatic_variants_for_sample_and_variant_caller.png: Visual representation showcasing somatic variants for each sample and variant caller.
summary_for_each_sample_and_variant_caller.tsv: Tabular summary offering detailed statistics for each sample and variant caller.
Let’s explore different scenarios for running Musta with various configurations using the musta detect command.
1. Run Musta with All Variant Callers:
This command runs Musta with all available variant callers:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--variant-file /path/to/variants.vcf \
--germline-resource /path/to/germline.vcf \
--bed-file /path/to/regions.bed.gz \
--dbsnp-file /path/to/dbsnp.vcf.gz \
2. Exclude Specific Variant Callers:
To exclude specific variant callers, use the --exclude options. For example, to exclude Mutect and Strelka:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--bed-file /path/to/regions.bed.gz \
--dbsnp-file /path/to/dbsnp.vcf.gz \
--exclude-mutect \
--exclude-strelka
3. Use Only Strict Variant Callers:
Run only the strict (restrictive) variant callers, which include Mutect, Lofreq, and Strelka:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--variant-file /path/to/variants.vcf \
--germline-resource /path/to/germline.vcf \
--bed-file /path/to/regions.bed.gz \
--dbsnp-file /path/to/dbsnp.vcf.gz \
--strict
4. Use Only Soft Variant Callers:
Run only the soft (permissive) variant callers, which include VarScan, Vardict, and Muse:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--bed-file /path/to/regions.bed,gz \
--dbsnp-file /path/to/dbsnp.vcf.gz \
--soft
5. Use Only Fast Variant Callers:
Run only the fast variant callers, which are Lofreq, VarScan, Strelka, and Muse:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--bed-file /path/to/regions.bed,gz \
--dbsnp-file /path/to/dbsnp.vcf.gz \
--fast
6. Run Only One Specific Variant Caller :
You can run only one specific variant caller using the --exclude- options,
in order to exclude all variant callers except the one you want to run.
For example, to run only the Mutect variant caller, you can use the following command:
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--variant-file /path/to/variants.vcf \
--germline-resource /path/to/germline.vcf \
--bed-file /path/to/regions.bed.gz \
--exclude-lofreq --exclude-strelka --exclude-muse --exclude-varscan --exclude-vardict
In this example, we’ve excluded all variant callers except “Mutect” by using the --exclude-lofreq,
--exclude-strelka, --exclude-muse, --exclude-varscan, and --exclude-vardict options.
Another way to run just one specific variant caller is by using a combination of filtering
(--strict, --fast, --soft) and --exclude- options.
For instance, to run only “Mutect,” you can use the --strict option to include Mutect and then exclude LoFreq and Strelka with --exclude-lofreq and --exclude-strelka.
musta detect \
--workdir /path/to/workdir \
--samples-file /path/to/samples.yml \
--reference-file /path/to/reference.fa \
--variant-file /path/to/variants.vcf \
--germline-resource /path/to/germline.vcf \
--bed-file /path/to/regions.bed.gz \
--strict \
--exclude-lofreq --exclude-strelka
These examples demonstrate different ways to configure Musta for somatic mutation detection, including using specific variant callers, excluding others, and selecting strict, soft, or fast variant callers based on your analysis needs.
In order to discuss the results obtained from the Musta Detection module, we didn’t utilize the “test-data-somatic” dataset for this evaluation.
Instead, we conducted our analysis on the original dataset, of which the demo serves as a representative subset: 23 tumor biopsies and a tumor-adjacent matched normal sample, all sequenced at an average depth of 74.4×. (See the dedicated section)
Let’s now start by looking at an extract from the summary file:
summary_for_each_sample_and_variant_caller.tsv| SAMPLE | VARIANT CALLER | TOTAL VARIANT COUNT | PASS VARIANT COUNT | RUNTIME (DAYS:HOURS:MINUTES:SECONDS) |
|---|---|---|---|---|
| patient_A05 | CONSENSUS | 7782 | 244 | 0:0:5:1 |
| patient_A05 | lofreq | 219 | 219 | 0:0:20:38 |
| patient_A05 | muse | 1655 | 957 | 0:1:32:53 |
| patient_A05 | mutect | 315 | 315 | 0:1:16:37 |
| patient_A05 | strelka | 9290 | 430 | 0:0:11:9 |
| patient_A05 | vardict | 70570 | 27820 | 1:0:23:2 |
| patient_A05 | varscan | 18345 | 16581 | 0:0:38:1 |
| patient_A25 | CONSENSUS | 3915 | 223 | 0:0:4:34 |
| patient_A25 | lofreq | 192 | 192 | 0:0:19:40 |
| patient_A25 | muse | 1674 | 945 | 0:1:33:12 |
| patient_A25 | mutect | 249 | 249 | 0:1:19:45 |
| patient_A25 | strelka | 4069 | 276 | 0:0:11:8 |
| patient_A25 | vardict | 63276 | 27532 | 1:0:2:51 |
| patient_A25 | varscan | 15039 | 13126 | 0:0:38:34 |
| patient_A58 | CONSENSUS | 5368 | 197 | 0:0:5:42 |
| patient_A58 | lofreq | 186 | 186 | 0:0:17:27 |
| patient_A58 | muse | 993 | 584 | 0:1:11:2 |
| patient_A58 | mutect | 355 | 355 | 0:1:11:31 |
| patient_A58 | strelka | 4817 | 323 | 0:0:11:8 |
| patient_A58 | vardict | 64383 | 27751 | 1:0:2:12 |
| patient_A58 | varscan | 13023 | 11379 | 0:0:34:13 |
| patient_A61 | CONSENSUS | 3311 | 201 | 0:0:4:11 |
| patient_A61 | lofreq | 170 | 170 | 0:0:16:26 |
| patient_A61 | muse | 1103 | 631 | 0:1:17:26 |
| patient_A61 | mutect | 253 | 253 | 0:1:14:13 |
| patient_A61 | strelka | 4232 | 296 | 0:0:10:38 |
| patient_A61 | vardict | 62143 | 27426 | 1:0:20:2 |
| patient_A61 | varscan | 11695 | 10097 | 0:0:34:2 |
| … | … | … | … | … |
These observations comply with the Filtering Options, highlighting the advantages of this approach over the utilization of all six variant callers simultaneously.
--strict: Runs only the restrictive variant callers, which include Mutect, LoFreq, and Strelka.--soft: Runs only the permissive variant callers, which include VarScan, VarDict, and MuSe.--fast: Executes only the fast variant callers, which are LoFreq, VarScan, and Strelka.The consensus strategy prioritizes variants identified by multiple callers, ensuring a more conservative selection. However, when integrating permissive and restrictive callers, there is a potential drawback: the rejection of a significant portion of variants called by the permissive ones.
Notably, the inclusion of VarDict, known for its extended runtime, alongside restrictive callers proves counterproductive and costly. This amalgamation does not contribute additional value and, in fact, may hinder efficiency.