Identification and visualization of antimicrobial resistance genes in a bacterial genome, including their genomic location and association with plasmids.
In this tutorial, we will demonstrate how to identify antimicrobial resistance (AMR) genes in a bacterial
genome using an assembled genome as input.
We will start from a bacterial genome assembly generated in
the previous tutorial.
Before starting the analysis, prepare your Galaxy workspace as follows:
Create a new Galaxy history and give it a meaningful name.
Import theShovill contigs dataset into the new history by dragging and dropping it from a previous history (see here for instructions on managing and copying datasets between histories).
Import the raw read filtered by fastp
To identify antimicrobial resistance (AMR) genes in assembled contigs,
tools such as ABRicate and StarAMR can be used.
In this tutorial, we focus on StarAMR, while ABRicate will be covered in a dedicated follow-up tutorial.
Staramr scans bacterial genome contigs against the ResFinder, PointFinder, and PlasmidFinder databases and generates a comprehensive summary report of the detected antimicrobial resistance genes.
StarAMR Tool with the following parameters:
There are 8 different output files produced by StarAMR
A summary of all detected AMR genes/mutations in each genome, one genome per line:
Isolate ID: The id of the isolate/genome file(s) passed to StarAMR
Quality Module: The isolate/genome file(s) pass/fail result(s) for the quality metrics
Genotype: The AMR genotype of the isolate.
Predicted Phenotype: The predicted AMR phenotype (drug resistances) for the isolate.
CGE Predicted Phenotype: The CGE-predicted AMR phenotype (drug resistances) for the isolate (CGE = Center for Genomic Epidemiology)
Plasmid: Plasmid types that were found for the isolate.
Scheme: The MLST scheme used
MLST stands for MultiLocus Sequence Typing. It is a technique for the typing of multiple loci, using DNA sequences of internal fragments of multiple housekeeping genes to characterize isolates of microbial species.
Here, StarAMR uses mlst to scan the contig files against traditional PubMLST typing schemes. The correspondance between the scheme and the bacteria genus and species is accessible in the map
Sequence Type: The sequence type that’s assigned when combining all allele types
Genome Length: The isolate/genome file(s) genome length(s)
N50 value: The isolate/genome file(s) N50 value(s)
Number of Contigs Greater Than Or Equal To 300 bp: The number of contigs greater or equal to 300 base pair in the isolate/genome file(s)
Quality Module Feedback: The isolate/genome file(s) detailed feedback for the quality metrics
A detailed summary of all detected AMR genes/mutations/plasmids/sequence type in each genome, one gene per line:
Isolate ID: The id of the isolate/genome file(s) passed to staramr.
Data: The particular gene detected from ResFinder, PlasmidFinder, PointFinder, or the sequence type.
Data Type: The type of gene (Resistance or Plasmid), or MLST.
Predicted Phenotype: The predicted AMR phenotype (drug resistances) found in ResFinder/PointFinder. Plasmids will be left blank by default.
CGE Predicted Phenotype: The CGE-predicted AMR phenotype (drug resistances) found in ResFinder/PointFinder. Plasmids will be left blank by default.
%Identity: The % identity of the top BLAST HSP to the gene.
%Overlap: THe % overlap of the top BLAST HSP to the gene (calculated as hsp length/total length * 100).
HSP Length/Total Length: The top BLAST HSP length over the gene total length (nucleotides).
Contig: The contig id containing this gene.
Start: The start of the gene (will be greater than End if on minus strand).
End: The end of the gene.
Accession: The accession of the gene from either ResFinder or PlasmidFinder database.
A tabular file of each AMR gene and additional BLAST information from the ResFinder database, one gene per line:
Isolate ID: The id of the isolate/genome file(s) passed to staramr.
Gene: The particular AMR gene detected.
Predicted Phenotype: The predicted AMR phenotype (drug resistances) for this gene.
CGE Predicted Phenotype: The CGE-predicted AMR phenotype (drug resistances) for this gene.
%Identity: The % identity of the top BLAST HSP to the AMR gene.
%Overlap: THe % overlap of the top BLAST HSP to the AMR gene (calculated as hsp length/total length * 100).
HSP Length/Total Length: The top BLAST HSP length over the AMR gene total length (nucleotides).
Contig: The contig id containing this AMR gene.
Start: The start of the AMR gene (will be greater than End if on minus strand).
End: The end of the AMR gene.
Accession: The accession of the AMR gene in the ResFinder database.
Sequence: The AMR Gene sequence
CGE Notes: Any CGE notes associated with the prediction
A tabular file of each AMR plasmid type and additional BLAST information from the PlasmidFinder database, one plasmid type per line:
Isolate ID: The id of the isolate/genome file(s) passed to staramr.
Plasmid: The particular plasmid type detected.
%Identity: The % identity of the top BLAST HSP to the plasmid type.
%Overlap: The % overlap of the top BLAST HSP to the plasmid type (calculated as hsp length/total length * 100).
HSP Length/Total Length: The top BLAST HSP length over the plasmid type total length (nucleotides).
Contig: The contig id containing this plasmid type.
Start: The start of the plasmid type (will be greater than End if on minus strand).
End: The end of the plasmid type.
Accession: The accession of the plasmid type in the PlasmidFinder database.
The command-line, database versions, and other settings used to run staramr.
An Excel spreadsheet containing the previous files as separate worksheets.
To get more information about the antibiotic resistant genes (ARG), we can check the CARD database (Comprehensive Antibiotic Resistance Database).
CARD can be very helpful to check all the resistance genes and check if it is logical to find the resistance gene in a specific bacteria.
Question
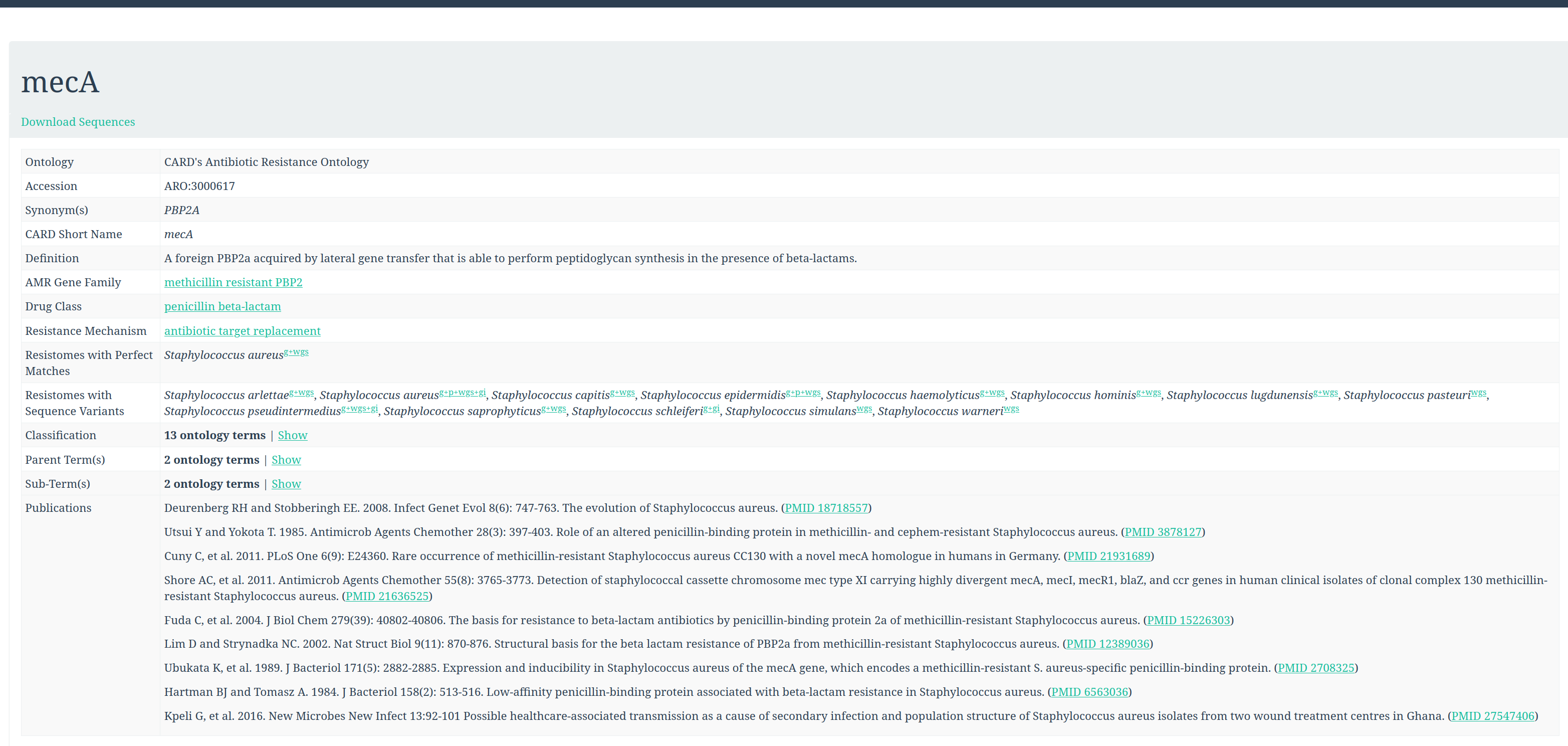
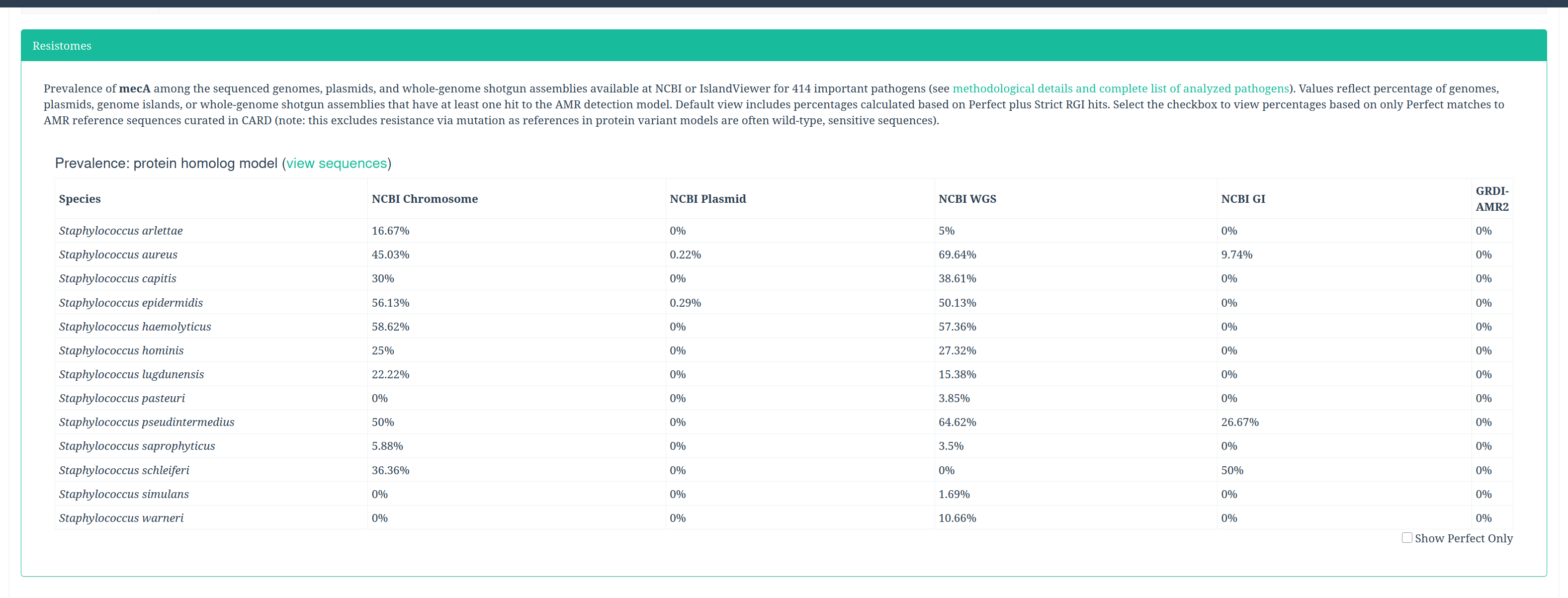
We would like to look at the ARGs and plasmid genes in their genomic context. To do that, we will use JBrowse
with several information:
Assembly as the reference
ARGs location
Contigs annotation (genes, etc)
Coverage of the contigs from the raw reads
The first step is to extract the location of the ARGs and plasmid genes on the contigs.
This information is available on the detailed_summary.tsv output of StarAMR.
The genes and their location are on the lines with a decimal value on column 6 or 7.
So to only get this information, we need to select lines with a decimal value (###.##) followed by a tab character,
the column separator in Galaxy. As a result, any lines without an identity or overlap value will be filtered out.
Select lines that match an expression with the following parameters:
This table can not be used directly in JBrowse. It first needs to be transformed in a standard format:
GFF3, a file format used for describing genes and other features of DNA, RNA and protein sequences
(see the previous tutorial for details).
Create a GFF file
Table to GFF3
Table: output of the above Select lines tool step
Source column or value: 3
Click on Run Tool
Question
In addition to antimicrobial resistance genes (ARGs) and plasmid-associated genes, it is useful to obtain functional information for all other genes present on the contigs.
For this purpose, we will use the genome annotation results generated in
the previous tutorial.
Specifically, drag and drop the GFF file produced by Bakta from the previous history into the current history,
as it contains the structural and functional annotation of the contigs.
To estimate the coverage of contigs and annotated genes,
the original sequencing reads are mapped back to the assembled contigs using Bowtie2.
Use the paired-end reads filtered with fastp,
imported by dragging and dropping them from
the assembly tutorial
history into the current history.
Bowtie2 with the following parameters:
MultiQC with the following parameters:
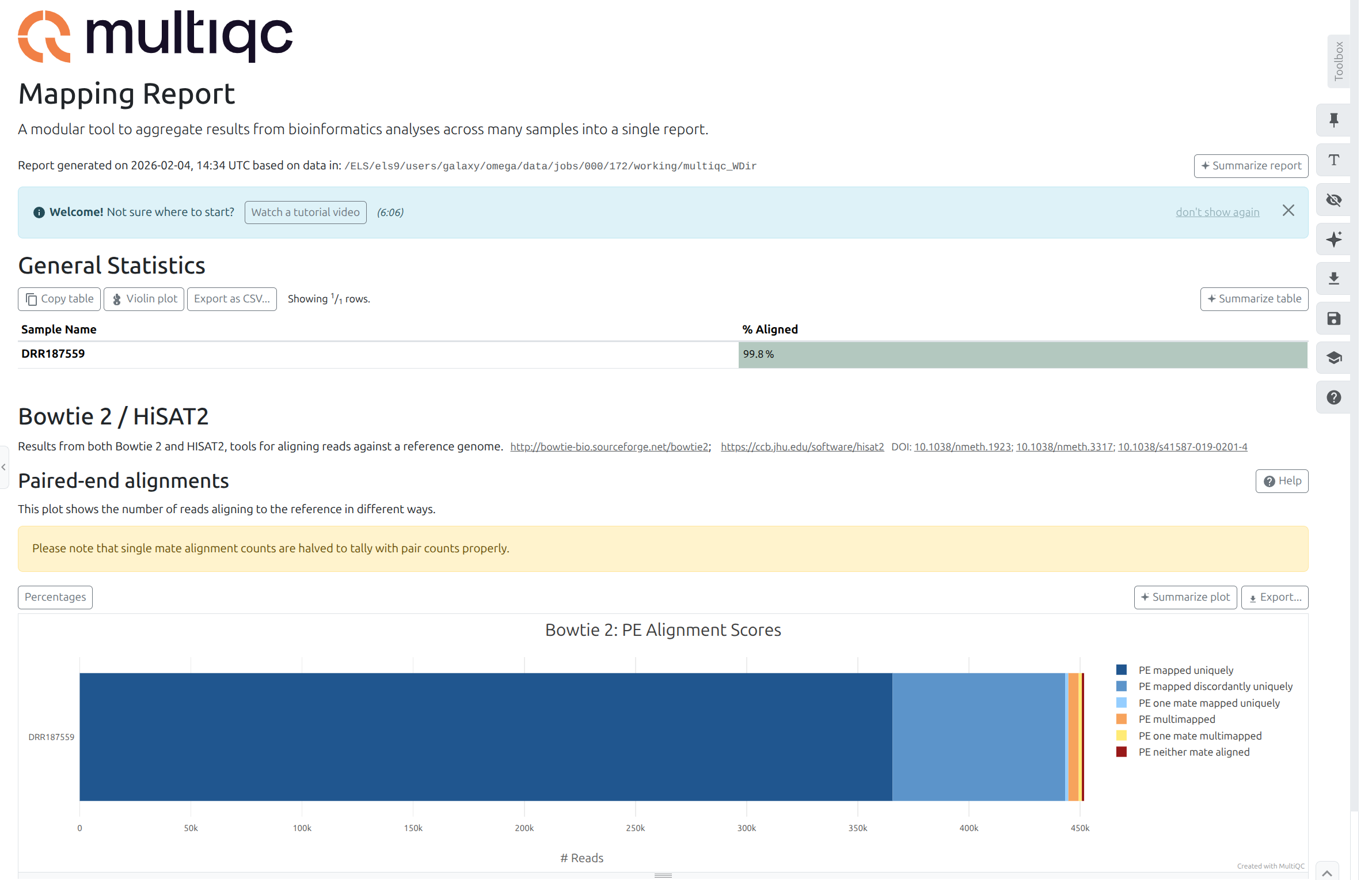
Question
We can now visualize the contigs, the mapping coverage, and the genes, using JBrowse and different information track.
JBrowse with the following parameters:
Bakta)JBrowseIn the output of the JBrowse you can view the mapped reads and the found genes against the reference genome.
With the search tools you can easily find genes of interest.
Using the Bowtie2 mapping output, low coverage regions can be detected.
This SNP detection can also give a clear view of where the data was less reliable
or where variations were located.
Question
What is the name for the gene found by Bakta corresponding to the rep16 gene found by StarAMR?
What are the NCBI Protein id and UniRef id for aac(6’)-aph(2’’)?
In this tutorial, contigs were scaned for AMR and plasmid genes. The genes were then visualized in their genomic context after contig annotation.