Bacterial genome assembly from Illumina MiSeq reads, covering data quality assessment, assembly generation, and assembly quality metrics.
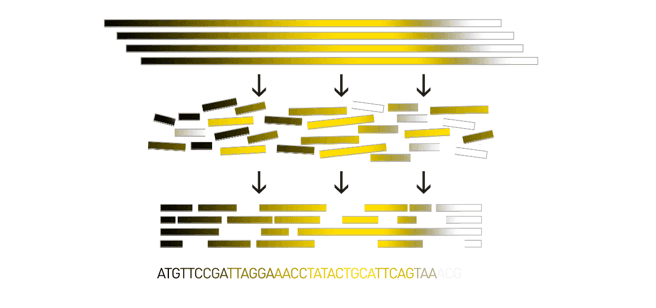
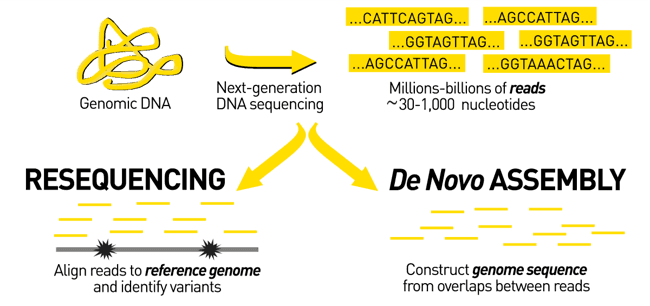
Sequencing produces short, unordered reads that represent only small fragments of the genome. Genome assembly addresses this limitation by reconstructing longer genomic sequences, enabling meaningful biological interpretation and downstream analyses such as genome annotation, resistance gene detection, and comparative genomics.
Genome assembly consists of aligning and reconstructing these fragments to form a continuous sequence (that of the chromosomes) or a set of contiguous sequences (called contigs or scaffolds)
Increases informational content
Assembled sequences (contigs/scaffolds) enable the analysis of complete genes, regulatory regions, and genomic context beyond single reads.
Enables reference-free analysis
In metagenomics and for many bacteria and viruses, suitable reference genomes may be unavailable or too distant.
Supports discovery of novel organisms and variants
Assembly allows reconstruction of genomes or genomic fragments not present in reference databases.
Improves functional annotation
Resistance genes, virulence factors, and viral proteins are more reliably identified on longer, continuous sequences.
Enables structural and comparative analyses
Including the study of plasmids, mobile genetic elements, genomic rearrangements, and strain-level comparisons.
Genome
Velvet, Velvet Optimizer, Spades, Abyss,
MIRA, Newbler, SGA, AllPaths, Ray, SOAPdenovo, …
Meta-genome
Meta Velvet, MetaSpades, SGA, custom scripts + above
Transcriptome
Trinity, Oases, Trans-abyss
And many, many others…
In this training we will build an assembly of a bacterial genome, from data produced in “Complete Genome Sequences of Eight Methicillin-Resistant Staphylococcus aureus Strains Isolated from Patients in Japan” Hikichi et al. 2019
Methicillin-resistant Staphylococcus aureus (MRSA) is a major pathogen causing nosocomial
infections, and the clinical manifestations of MRSA range from asymptomatic colonization of the nasal mucosa to soft tissue infection to fulminant invasive disease.
NOTE! Paired-end reads were generated using a MiSeq reagent kit (v3-600) on the MiSeq platform (Illumina).
https://zenodo.org/record/10669812/files/DRR187559_1.fastqsanger.bz2
https://zenodo.org/record/10669812/files/DRR187559_2.fastqsanger.bz2
.fastqsanger.bz2 and keep only the sequence run ID (DRR187559_1 and DRR187559_2)
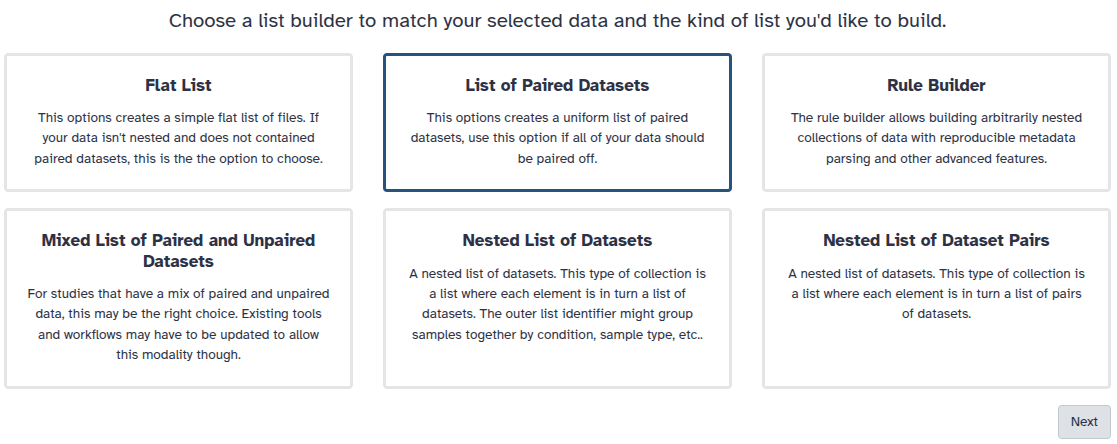
_1 and _2 or _R1 and _R2. Click on Next at the bottom.
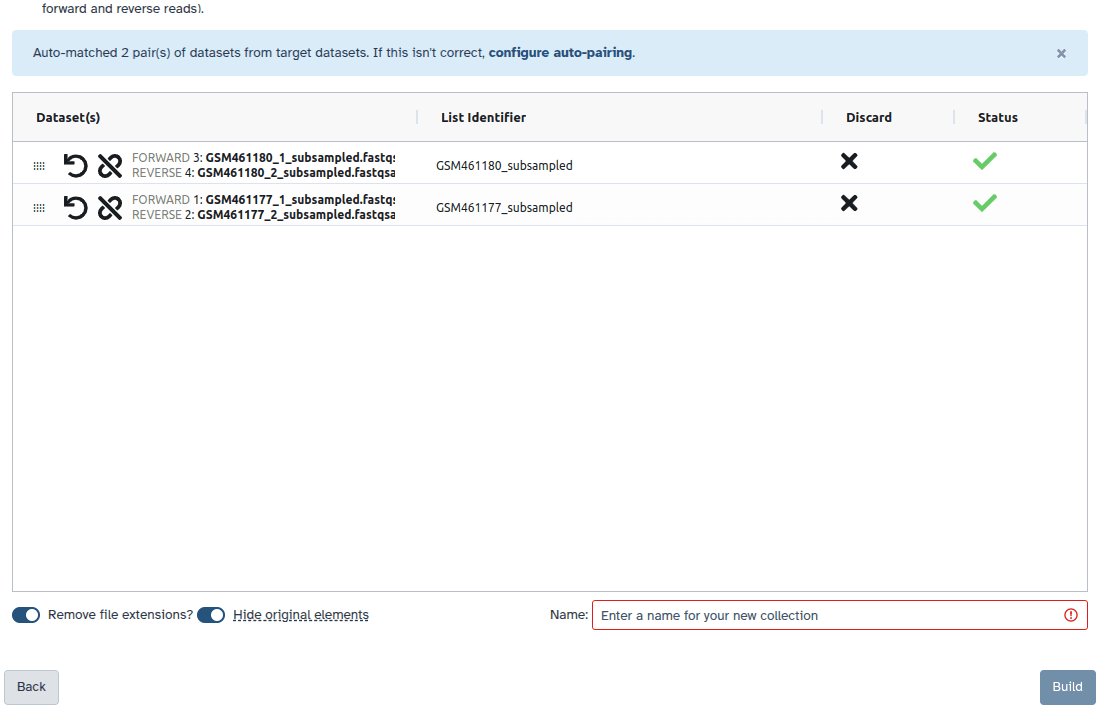
Before performing any genome assembly, it is essential to assess the quality of the input reads. Key questions to consider include:
What is the sequencing coverage of the genome?
What is the overall quality of the reads?
Is additional sequencing required?
Are the data suitable for the intended downstream analyses?
In the previous tutorial, we used FastQC for read quality assessment.
In this lesson, we will use Falco, an efficiency-optimized reimplementation of FastQC, designed to provide the same quality metrics with improved performance.
Falco Tool with the following parameters

|

|
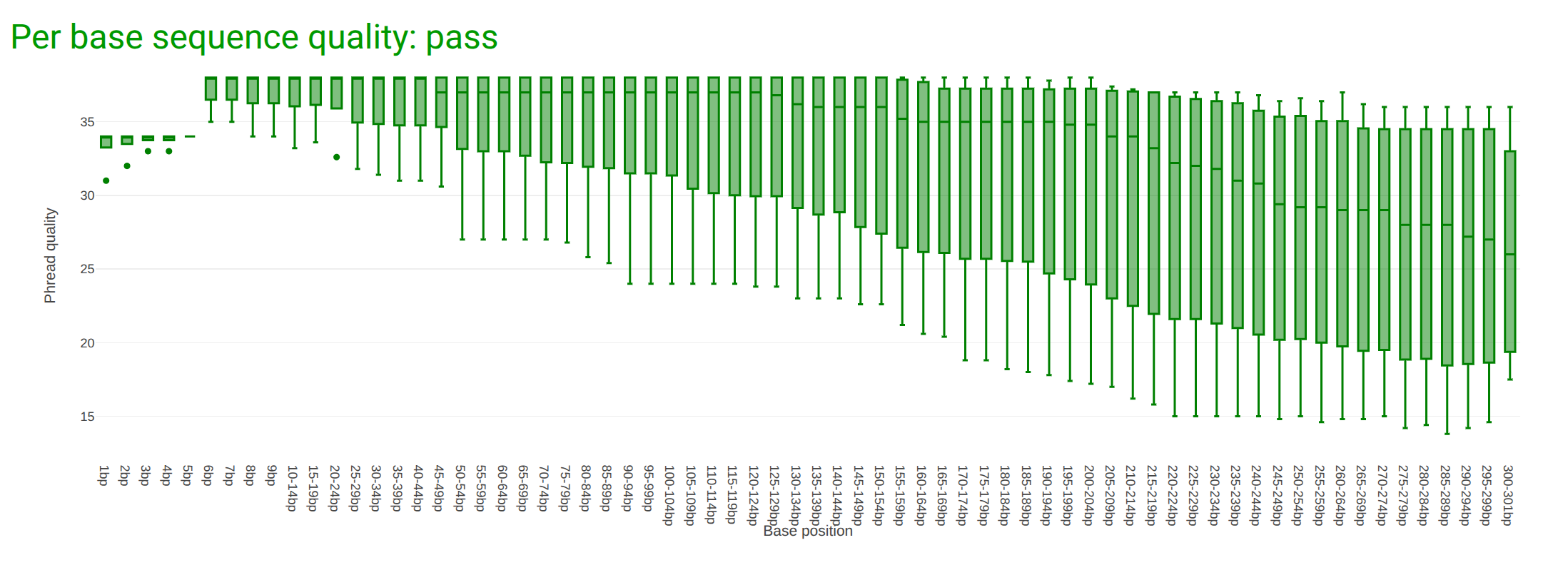
Depending on the analysis it could be possible that a certain quality or length is needed.
In this case we are going to trim the data using fastp
(or Trim-Galore etc.)
fastp with the following parameters:
fastp generates also a report, similar to Falco, useful to compare the impact of the trimming and filtering.
Question
Exercises
MultiQC on the JSON output generated by fastp.Before proceeding with genome assembly, it is important to check if the expected species or strains can be identified in the data or if there is any contamination.
As this step is not specific to genome assembly, it is covered in a dedicated tutorial.
Now that the quality of the reads is determined and the data is filtered and/or trimmed, we can try to assemble the reads together to build longer sequences.
There are many tools that create assembly for short-read data, e.g. SPAdes,
Abyss or Velvet.
In this tutorial, we use Shovill.
Shovill is a SPAdes-based genome assembler, improved to work faster and only for smaller (bacterial) genomes.
Shovill is for isolate data only, primarily small haploid organisms. It will NOT work on metagenomes or larger genomes
Shovill with the following parameters:
Shovill estimates it during pre-processing.
Nonetheless, providing it manually is recommended for more robust results with ‘heavy’ samples.Question
Individual Assembly: Reads from each sample are assembled independently. This approach is ideal for preserving strain-specific variations and is computationally efficient. However, it may fail to reconstruct low-abundance sequences that lack sufficient coverage within a single library.
Co-assembly: Reads from multiple samples are pooled and assembled together into a single set of contigs. This increases the overall sequencing depth, facilitating the recovery of rare taxa. The main drawbacks include high computational requirements and the potential for generating chimeric contigs due to microdiversity between samples.
| Situation | Recommended Choice | Reason |
|---|---|---|
| Pure bacterial isolates | Individual Assembly | Better for preserving specific strain identity. |
| Metagenomics (same site/patient) | Co-Assembly | Increases coverage to recover rare species. |
| Metagenomics (very different sites) | Individual Assembly | Avoids creating chimeras from unrelated species. |
| Limited computing resources (RAM) | Individual Assembly | Requires significantly less memory per run. |
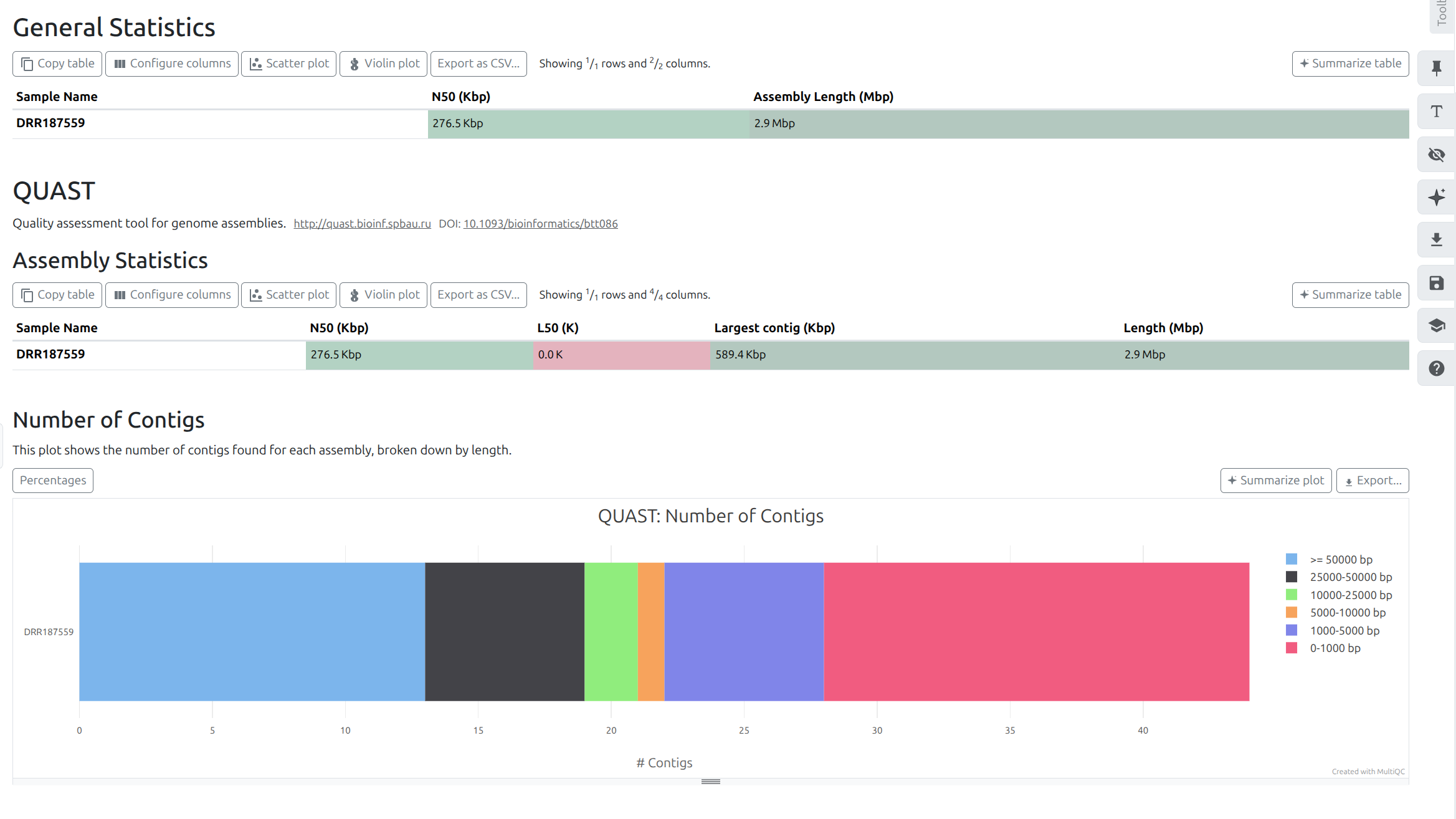
Quast Tool with parameters
Shovillfastp output 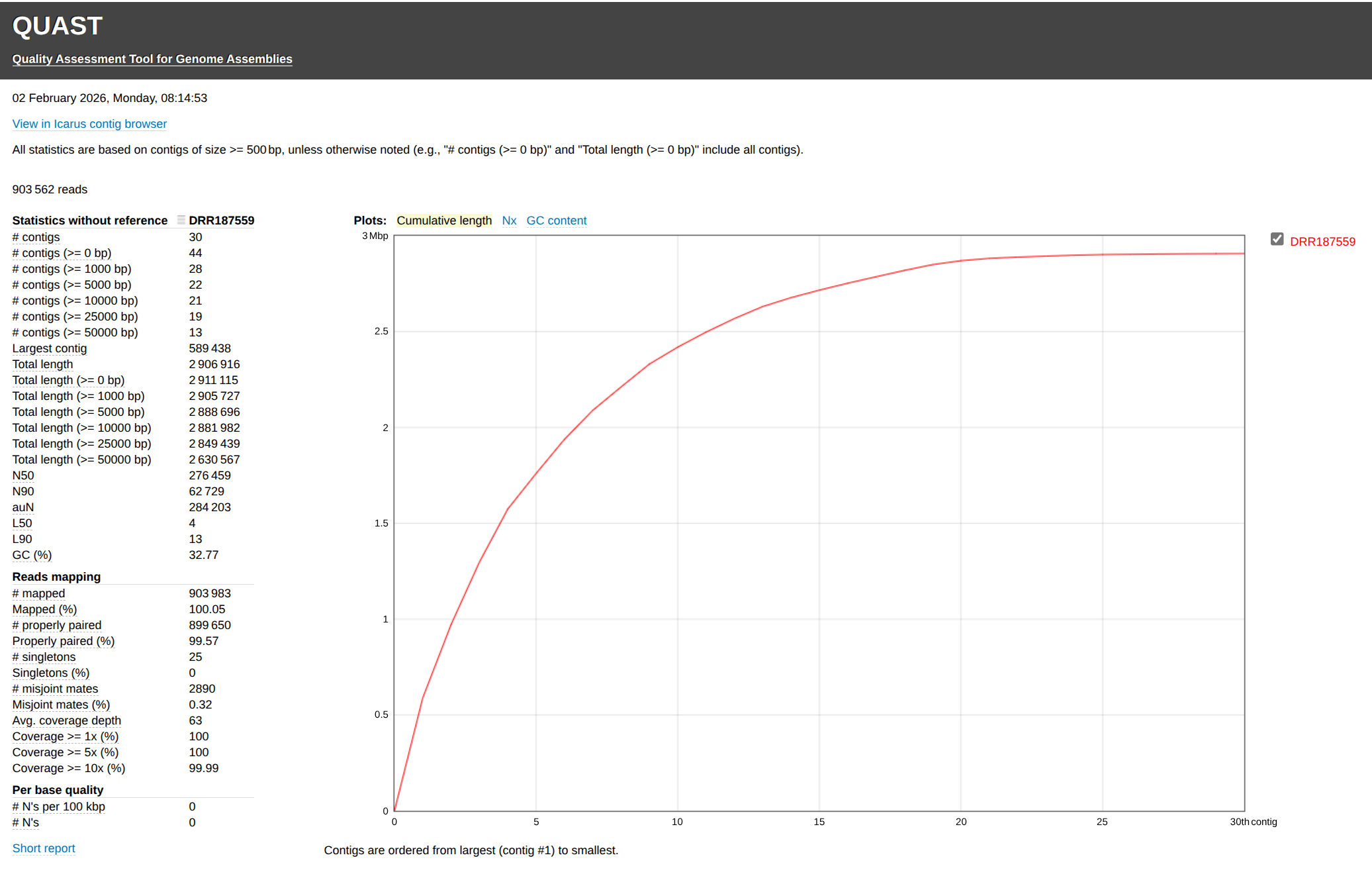
The following metrics are the most critical indicators of a high-quality bacterial assembly:
N50 - The N50 value indicates that 50% of the total assembly length is contained in contigs of this size or larger.
Significance: It measures the continuity of the assembly.
Quality Benchmark: For a bacterial isolate, an N50 > 100–200 kb is generally considered excellent. A low N50 (e.g., < 20 kb) suggests a highly fragmented assembly.
Number of Contigs (# contigs) - This represents the total number of fragments the genome has been broken into.
Significance: It measures fragmentation.
Quality Benchmark: Ideally, this should be below 100-150 for a single bacterial genome. Thousands of contigs often indicate issues with repeats or insufficient sequencing depth.
Total Length - The cumulative length of all contigs should be close to the expected genome size (e.g., ~2.9 Mb for MRSA).
Significance: It measures completeness.
Quality Benchmark: The length should be within ±5-10% of the reference genome. Significant deviations may suggest contamination (if too high) or missing genomic regions (if too low).
GC Content (%) - The percentage of Guanine and Cytosine bases in the assembly.
Significance: It serves as a check for taxonomic identity and contamination.
Quality Benchmark: It must be consistent with the target species (e.g., ~32.89% for MRSA).
See Quast Manual for more details.
Question
MultiQC tool with following parameters
You are required to repeat the Assembly + QUAST workflow using SPAdes (with --isolate optiopn)
and compare the results with your previous Shovill assembly.

In this tutorial, we prepared short reads, assembled them, and inspect the produced assembly for its quality. The assembly, even if uncomplete, is reasonable good to be used in downstream analysis: